Identification of a Novel Missense Variant in SPDL1 Associated with Idiopathic Pulmonary Fibrosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BLOC-1 and BLOC-3 Regulate VAMP7 Cycling to and From
BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers Megan K. Dennis1,2, Cédric Delevoye3,4, Amanda Acosta-Ruiz1,2, Ilse Hurbain3,4, Maryse Romao3,4, Geoffrey G. Hesketh5, Philip S. Goff6, Elena V. Sviderskaya6, Dorothy C. Bennett6, J. Paul Luzio5, Thierry Galli7, David J. Owen5, Graça Raposo3,4 and Michael S. Marks1,2,8 1Dept. of Pathology and Laboratory Medicine, Children's Hospital of Philadelphia Research Institute, and 2Dept. of Pathology and Laboratory Medicine and Dept of Physiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA; 3Institut Curie, PSL Research University, CNRS, UMR144, Structure and Membrane Compartments, F-75005, Paris, France; 4 Institut Curie, PSL Research University, CNRS, UMR144, Cell and Tissue Imaging Facility (PICT-IBiSA), F-75005, Paris, France; 5Cambridge Institute for Medical Research, University of Cambridge, Cambridge, UK; 6Cell Biology & Genetics Research Centre, St. George’s , University of London, London, UK; 7Sorbonne Paris-Cité, Univ. Paris-Diderot, Institut Jacques Monod, CNRS UMR7592, INSERM ERL U950, Membrane Traffic in Health & Disease, Paris, France. 8To whom correspondence should be addressed: Michael S. Marks, Ph.D. Dept. of Pathology & Laboratory Medicine Children's Hospital of Philadelphia Research Institute 816G Abramson Research Center 3615 Civic Center Blvd. Philadelphia, PA 19104 Tel: 215-590-3664 Email: [email protected] Running title: VAMP7 into and out of melanosomes Keywords: SNARE, lysosome-related organelle, melanogenesis, Hermansky-Pudlak syndrome, recycling, endosome, 2 ABSTRACT Endomembrane organelle maturation requires cargo delivery via fusion with membrane transport intermediates and recycling of fusion factors to their sites of origin. -

Diagnosing Platelet Secretion Disorders: Examples Cases
Diagnosing platelet secretion disorders: examples cases Martina Daly Department of Infection, Immunity and Cardiovascular Disease, University of Sheffield Disclosures for Martina Daly In compliance with COI policy, ISTH requires the following disclosures to the session audience: Research Support/P.I. No relevant conflicts of interest to declare Employee No relevant conflicts of interest to declare Consultant No relevant conflicts of interest to declare Major Stockholder No relevant conflicts of interest to declare Speakers Bureau No relevant conflicts of interest to declare Honoraria No relevant conflicts of interest to declare Scientific Advisory No relevant conflicts of interest to declare Board Platelet granule release Agonists (FIIa, Collagen, ADP) Signals Activation Shape change Membrane fusion Release of granule contents Platelet storage organelles lysosomes a granules Enzymes including cathepsins Adhesive proteins acid hydrolases Clotting factors and their inhibitors Fibrinolytic factors and their inhibitors Proteases and antiproteases Growth and mitogenic factors Chemokines, cytokines Anti-microbial proteins Membrane glycoproteins dense (d) granules ADP/ATP Serotonin histamine inorganic polyphosphate Platelet a-granule contents Type Prominent components Membrane glycoproteins GPIb, aIIbb3, GPVI Clotting factors VWF, FV, FXI, FII, Fibrinogen, HMWK, FXIII? Clotting inhibitors TFPI, protein S, protease nexin-2 Fibrinolysis components PAI-1, TAFI, a2-antiplasmin, plasminogen, uPA Other protease inhibitors a1-antitrypsin, a2-macroglobulin -

A Network of Bhlhzip Transcription Factors in Melanoma: Interactions of MITF, TFEB and TFE3
A network of bHLHZip transcription factors in melanoma: Interactions of MITF, TFEB and TFE3 Josué A. Ballesteros Álvarez Thesis for the degree of Philosophiae Doctor January 2019 Net bHLHZip umritunarþátta í sortuæxlum: Samstarf milli MITF, TFEB og TFE3 Josué A. Ballesteros Álvarez Ritgerð til doktorsgráðu Leiðbeinandi/leiðbeinendur: Eiríkur Steingrímsson Doktorsnefnd: Margrét H. Ögmundsdóttir Þórarinn Guðjónsson Jórunn E. Eyfjörð Lars Rönnstrand Janúar 2019 Thesis for a doctoral degree at tHe University of Iceland. All rigHts reserved. No Part of tHis Publication may be reProduced in any form witHout tHe Prior permission of the copyright holder. © Josue A. Ballesteros Álvarez. 2019 ISBN 978-9935-9421-4-2 Printing by HáskólaPrent Reykjavik, Iceland 2019 Ágrip StjórnPróteinin MITF , TFEB, TFE3 og TFEC (stundum nefnd MiT-TFE þættirnir) tilheyra bHLHZip fjölskyldu umritunarþátta sem bindast DNA og stjórna tjáningu gena. MITF er mikilvægt fyrir myndun og starfsemi litfruma en ættingjar þess, TFEB og TFE3, stjórna myndun og starfsemi lysósóma og sjálfsáti. Sjálfsát er líffræðilegt ferli sem gegnir mikilvægu hlutverki í starfsemi fruma en getur einnig haft áHrif á myndun og meðHöndlun sjúkdóma. Í verkefni þessu var samstarf MITF, TFE3 og TFEB Próteinanna skoðað í sortuæxlisfrumum og hvaða áhrif þau Hafa á tjáningu hvers annars. Eins og MITF eru TFEB og TFE3 genin tjáð í sortuæxlisfrumum og sortuæxlum; TFEC er ekki tjáð í þessum frumum og var því ekki skoðað í þessu verkefni. Með notkun sérvirkra hindra var sýnt að boðleiðir hafa áhrif á staðsetningu próteinanna þriggja í sortuæxlisfrumum. Umritunarþættir þessir geta bundist skyldum DNA-bindisetum og haft áhrif á tjáningu gena sem eru nauðsynleg fyrir myndun bæði lýsósóma og melanósóma. -

Supplementary Tables
Supplementary Tables Supplementary Table S1: Preselected miRNAs used in feature selection Univariate Cox proportional hazards regression analysis of the endpoint freedom from recurrence in the training set (DKTK-ROG sample) allowed the pre-selection of 524 miRNAs (P< 0.5), which were used in the feature selection. P-value was derived from log-rank test. miRNA p-value miRNA p-value miRNA p-value miRNA p-value hsa-let-7g-3p 0.0001520 hsa-miR-1304-3p 0.0490161 hsa-miR-7108-5p 0.1263245 hsa-miR-6865-5p 0.2073121 hsa-miR-6825-3p 0.0004257 hsa-miR-4298 0.0506194 hsa-miR-4453 0.1270967 hsa-miR-6893-5p 0.2120664 hsa-miR-668-3p 0.0005188 hsa-miR-484 0.0518625 hsa-miR-200a-5p 0.1276345 hsa-miR-25-3p 0.2123829 hsa-miR-3622b-3p 0.0005885 hsa-miR-6851-3p 0.0531446 hsa-miR-6090 0.1278692 hsa-miR-3189-5p 0.2136060 hsa-miR-6885-3p 0.0006452 hsa-miR-1276 0.0557418 hsa-miR-148b-3p 0.1279811 hsa-miR-6073 0.2139702 hsa-miR-6875-3p 0.0008188 hsa-miR-3173-3p 0.0559962 hsa-miR-4425 0.1288330 hsa-miR-765 0.2141536 hsa-miR-487b-5p 0.0011381 hsa-miR-650 0.0564616 hsa-miR-6798-3p 0.1293342 hsa-miR-338-5p 0.2153079 hsa-miR-210-5p 0.0012316 hsa-miR-6133 0.0571407 hsa-miR-4472 0.1300006 hsa-miR-6806-5p 0.2173515 hsa-miR-1470 0.0012822 hsa-miR-4701-5p 0.0571720 hsa-miR-4465 0.1304841 hsa-miR-98-5p 0.2184947 hsa-miR-6890-3p 0.0016539 hsa-miR-202-3p 0.0575741 hsa-miR-514b-5p 0.1308790 hsa-miR-500a-3p 0.2185577 hsa-miR-6511b-3p 0.0017165 hsa-miR-4733-5p 0.0616138 hsa-miR-378c 0.1317442 hsa-miR-4515 0.2187539 hsa-miR-7109-3p 0.0021381 hsa-miR-595 0.0629350 hsa-miR-3121-3p -

Genomic Aberrations Associated with Erlotinib Resistance in Non-Small Cell Lung Cancer Cells
ANTICANCER RESEARCH 33: 5223-5234 (2013) Genomic Aberrations Associated with Erlotinib Resistance in Non-small Cell Lung Cancer Cells MASAKUNI SERIZAWA1, TOSHIAKI TAKAHASHI2, NOBUYUKI YAMAMOTO2,3 and YASUHIRO KOH1 1Drug Discovery and Development Division, Shizuoka Cancer Center Research Institute, Sunto-gun, Shizuoka, Japan; 2Division of Thoracic Oncology, Shizuoka Cancer Center Hospital, Sunto-gun, Shizuoka, Japan; 3Third Department of Internal Medicine, Wakayama Medical University, Kimiidera, Wakayama, Japan Abstract. Background/Aim: Mechanisms of resistance to mutations develop resistance, usually within one year of epidermal growth factor receptor (EGFR)-tyrosine kinase treatment. Therefore, there is an urgent need to elucidate the inhibitors (TKIs) in non-small cell lung cancer (NSCLC) underlying mechanisms of resistance in such tumors to are not fully-understood. In this study we aimed to overcome this obstacle (11-14, 17, 24). Recent studies elucidate remaining unknown mechanisms using erlotinib- suggest that mechanisms of acquired resistance to EGFR- resistant NSCLC cells. Materials and Methods: We TKIs can be categorized into three groups: occurrence of performed array comparative genomic hybridization genetic alterations, activation of downstream pathways via (aCGH) to identify genomic aberrations associated with bypass signaling, and phenotypic transformation (15, 16, 21); EGFR-TKI resistance in erlotinib-resistant PC-9ER cells. therapeutic strategies to overcome these resistance Real-time polymerase chain reaction (PCR) and mechanisms are under development. However, although the immunoblot analyses were performed to confirm the results causes of acquired resistance to EGFR-TKIs have been of aCGH. Results: Among the five regions with copy investigated, in more than 30% of patients with acquired number gain detected in PC-9ER cells, we focused on resistance to EGFR-TKI treatment, the mechanisms remain 22q11.2-q12.1 including v-crk avian sarcoma virus CT10 unknown (15). -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -
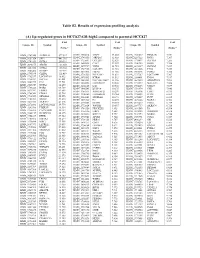
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -

Mutation of Melanosome Protein RAB38 in Chocolate Mice
Mutation of melanosome protein RAB38 in chocolate mice Stacie K. Loftus*, Denise M. Larson*, Laura L. Baxter*, Anthony Antonellis*†, Yidong Chen‡, Xufeng Wu§, Yuan Jiang‡, Michael Bittner‡, John A. Hammer III§, and William J. Pavan*¶ *Genetic Disease Research Branch and ‡Cancer Genetics Branch, National Human Genome Research Institute, §Laboratory of Cell Biology, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892; and †Graduate Genetics Program, George Washington University, Washington, DC 20052 Communicated by Francis S. Collins, National Institutes of Health, Bethesda, MD, February 13, 2002 (received for review January 2, 2002) Mutations of genes needed for melanocyte function can result in crest-derived and other control cell lines. Clustering of the oculocutaneous albinism. Examination of similarities in human resulting expression profiles provided a powerful way to organize gene expression patterns by using microarray analysis reveals that the common patterns found among thousands of gene expression RAB38, a small GTP binding protein, demonstrates a similar ex- measurements and identify genes with similar distinctive expres- pression profile to melanocytic genes. Comparative genomic anal- sion patterns among the experimental samples (6). Analysis of ysis localizes human RAB38 to the mouse chocolate (cht) locus. A genes contained within a cluster has revealed that these genes are G146T mutation occurs in the conserved GTP binding domain of often functionally related within the cell (7, 8). Using this RAB38 in cht mice. Rab38cht͞Rab38cht mice exhibit a brown coat approach we identified genes clustered with known pigmenta- similar in color to mice with a mutation in tyrosinase-related tion genes, thereby categorizing RAB38 as a candidate pigmen- protein 1 (Tyrp1), a mouse model for oculocutaneous albinism. -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Hermansky-Pudlak Syndrome
Chapter 8 Hermansky-Pudlak Syndrome Naoki Oiso and Akira Kawada Additional information is available at the end of the chapter http://dx.doi.org/10.5772/53573 1. Introduction Oculocutaneous albinism is classified into non-syndromic oculocutaneous albinism (OCA) and syndromic OCA including Hermansky-Pudlak syndrome (HPS), Chediak-Higashi syndrome (CHS) and Griscelli syndrome (GS). Both non-syndromic and syndromic OCAs are autosomal recessive disorders. Human HPS is genetically divided into nine forms, HPS type 1 (HPS-1) to HPS-9. Human HPS can be sub-classified into four subgroups which are associated with protein complexes encoded by the causative genes. In this session, we summarize (1) the clinical features of HPS, (2) the mice and rat models of HPS, and (3) the molecular functions. 2. The clinical features of HPS In 1959, Hermansky and Pudklak described two cases of OCA associated with hemorrhagic diathesis.1 Currently, the condition is known as HPS. HPS is a rare heterogeneous autosomal recessive syndrome which is typically characterized by OCA, bleeding diathesis, and lysosomal ceroid storage resulting from defects of multiple cytoplasmic organelles: melanosomes, platelet dense core granules, and lysosomes.2 The storage of ceroid-like material in lysosomes induces restrictive lung disease, ulcerative colitis, kidney failure, and cardiomyopathy. Accumulation of mice models, identification of causative genes and functional analysis indicated that HPS could be sub-classified into four groups according to four protein complexes, biogenesis of lysosome-related organelles complex-3 (BLOC-3) (HPS-1 and HPS- 4), adaptor protein-3 (AP-3) (HPS-2), BLOC-2 (HPS-3, HPS-5 and HPS-6) and BLOC-1( HPS- 7, HPS-8 and HPS-9).3-5 Currently, more than 16 mice strains and more than 2 rat strains are known as models of human HPS (Table 1). -

NIH Public Access Author Manuscript Annu Rev Genomics Hum Genet
NIH Public Access Author Manuscript Annu Rev Genomics Hum Genet. Author manuscript; available in PMC 2009 October 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Annu Rev Genomics Hum Genet. 2008 ; 9: 359±386. doi:10.1146/annurev.genom.9.081307.164303. Disorders of Lysosome-related Organelle Biogenesis: Clinical and Molecular Genetics Marjan Huizing1, Amanda Helip-Wooley2, Wendy Westbroek2, Meral Gunay-Aygun2, and William A. Gahl2 Marjan Huizing: [email protected]; Amanda Helip-Wooley: [email protected]; Wendy Westbroek: [email protected]; Meral Gunay-Aygun: [email protected]; William A. Gahl: [email protected] 1 Cell Biology of Metabolic Disorders Unit, National Institutes of Health, Bethesda, Maryland 20892 2 Section on Human Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892 Abstract Lysosome-related organelles (LROs) are a heterogeneous group of vesicles that share various features with lysosomes, but are distinct in function, morphology, and composition. The biogenesis of LROs employs a common machinery, and genetic defects in this machinery can affect all LROs or only an individual LRO, resulting in a variety of clinical features. In this review, we discuss the main components in LRO biogenesis. We also address the function, composition and resident cell type of the major LROs. Finally, we describe the clinical characteristics of the major human LRO disorders. Keywords Chediak-Higashi syndrome; Griscelli syndrome; Hermansky-Pudlak syndrome; melanosome; platelet INTRODUCTION Lysosomes are membrane-bound cytoplasmic organelles that serve as major degradative compartments in eukaryotic cells (65).