BCL6 Repression of EP300 in Human Diffuse Large B Cell Lymphoma Cells Provides a Basis for Rational Combinatorial Therapy Leandro C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

RNA-Binding Protein Hnrnpll Regulates Mrna Splicing and Stability During B-Cell to Plasma-Cell Differentiation
RNA-binding protein hnRNPLL regulates mRNA splicing and stability during B-cell to plasma-cell differentiation Xing Changa,b, Bin Lic, and Anjana Raoa,b,d,e,1 Divisions of aSignaling and Gene Expression and cVaccine Discovery, La Jolla Institute for Allergy and Immunology, La Jolla, CA 92037; bSanford Consortium for Regenerative Medicine, La Jolla, CA 92037; and dDepartment of Pharmacology and eMoores Cancer Center, University of California at San Diego, La Jolla, CA 92093 Contributed by Anjana Rao, December 2, 2014 (sent for review July 20, 2014) Posttranscriptional regulation is a major mechanism to rewire the RBP-binding sites, thus validating the specificity of RBP binding transcriptomes during differentiation. Heterogeneous nuclear to coprecipitating RNAs and mapping RBP-binding sites on the RNA-binding protein LL (hnRNPLL) is specifically induced in terminally validated RNAs at close to single-nucleotide resolution (8). differentiated lymphocytes, including effector T cells and plasma Heterogeneous nuclear RNA-binding proteins (hnRNPs) is cells. To study the molecular functions of hnRNPLL at a genome- the term applied to a collection of unrelated nuclear RBPs. wide level, we identified hnRNPLL RNA targets and binding sites in hnRNPLL was identified through a targeted lentiviral shRNA plasma cells through integrated Photoactivatable-Ribonucleoside- screen for regulators of CD45RA to CD45RO switching during Enhanced Cross-Linking and Immunoprecipitation (PAR-CLIP) and memory T-cell development (9) and independently through RNA sequencing. hnRNPLL preferentially recognizes CA dinucleo- two separate screens performed by different groups for exclusion tide-containing sequences in introns and 3′ untranslated regions of CD45 exon 4 in a minigene context (10) and for altered CD44 (UTRs), promotes exon inclusion or exclusion in a context-dependent and CD45R expression on T cells in N-ethyl-N-nitrosourea manner, and stabilizes mRNA when associated with 3′ UTRs. -

TGF-Β1 Signaling Targets Metastasis-Associated Protein 1, a New Effector in Epithelial Cells
Oncogene (2011) 30, 2230–2241 & 2011 Macmillan Publishers Limited All rights reserved 0950-9232/11 www.nature.com/onc ORIGINAL ARTICLE TGF-b1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells SB Pakala1, K Singh1,3, SDN Reddy1, K Ohshiro1, D-Q Li1, L Mishra2 and R Kumar1 1Department of Biochemistry and Molecular Biology and Institute of Coregulator Biology, The George Washington University Medical Center, Washington, DC, USA and 2Department of Gastroenterology, Hepatology and Nutrition, The University of Texas MD Anderson Cancer Center, Houston, TX, USA In spite of a large number of transforming growth factor b1 gene chromatin in response to upstream signals. The (TGF-b1)-regulated genes, the nature of its targets with TGF-b1-signaling is largely mediated by Smad proteins roles in transformation continues to be poorly understood. (Massague et al., 2005) where Smad2 and Smad3 are Here, we discovered that TGF-b1 stimulates transcription phosphorylated by TGF-b1-receptors and associate with of metastasis-associated protein 1 (MTA1), a dual master the common mediator Smad4, which translocates to the coregulator, in epithelial cells, and that MTA1 status is a nucleus to participate in the expression of TGF-b1-target determinant of TGF-b1-induced epithelial-to-mesenchymal genes (Deckers et al., 2006). Previous studies have shown transition (EMT) phenotypes. In addition, we found that that CUTL1, also known as CDP (CCAAT displacement MTA1/polymerase II/activator protein-1 (AP-1) co-activator protein), a target of TGF-b1, is needed for its short-term complex interacts with the FosB-gene chromatin and stimu- effects of TGF-b1 on cell motility involving Smad4- lates its transcription, and FosB in turn, utilizes FosB/histone dependent pathway (Michl et al.,2005). -

Cross-Family Dimerization of Transcription Factors Fos/Jun
Proc. Nati. Acad. Sci. USA Vol. 88, pp. 3720-3724, May 9, 1991 Biochemistry Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity (gene regulation/oncogenes/leucine zipper/protein dimerization/transcription) TSONWIN HAI*t AND ToM CURRANt of Medical Biochemistry and Ohio State Biotechnology Center, Ohio State University, 1060 Carmack Road, Columbus, OH 43210; and tDepartment*Department of Molecular Oncology and Virology, Roche Institute of Molecular Biology, Roche Research Center, Nutley, NJ 07110 Communicated by Herbert Weissbach, January 31, 1991 ABSTRACT The Fos/Jun and ATF/CREB families of Table 1. Mammalian Fos/Jun and ATF/CREB families transcription factors function in coupling extracellular signals Family Subfamily Gene(s) to alterations in expression of specific target genes. Like many eukaryotic transcription factors, these proteins bind to DNA as Fos/Jun Fos fos, fra-), fra-2, fosB dimers. Dimerization is mediated by a structure known as the Jun c-jun, junB, junD "leucine-zipper" motif. Although Fos/Jun and ATF/CREB ATF/CREB CREB CREB, ATF-I were previously thought to interact preferentially with differ- CRE-BP1 CRE-BPI, ATF-a ent DNA regulatory elements (the AP-1/TRE and ATF/CRE ATF-3 ATF-3* sites, respectively), we rind that members of these two families ATF-4 ATF4* form selective cross-family heterodimers. The resulting het- Alternative names for CRE-BPI are ATF-2 and HB16. ACREB is erodimers display distinguishable DNA binding speclfcities a spliced variant ofCREB, ATF-aA is a spliced variant ofATF-a, and from each other and from their parental homodimers. -

Neurobiological Functions of the Period Circadian Clock 2 Gene, Per2
Review Biomol Ther 26(4), 358-367 (2018) Neurobiological Functions of the Period Circadian Clock 2 Gene, Per2 Mikyung Kim, June Bryan de la Peña, Jae Hoon Cheong and Hee Jin Kim* Department of Pharmacy, Uimyung Research Institute for Neuroscience, Sahmyook University, Seoul 01795, Republic of Korea Abstract Most organisms have adapted to a circadian rhythm that follows a roughly 24-hour cycle, which is modulated by both internal (clock-related genes) and external (environment) factors. In such organisms, the central nervous system (CNS) is influenced by the circadian rhythm of individual cells. Furthermore, the period circadian clock 2 (Per2) gene is an important component of the circadian clock, which modulates the circadian rhythm. Per2 is mainly expressed in the suprachiasmatic nucleus (SCN) of the hypothalamus as well as other brain areas, including the midbrain and forebrain. This indicates that Per2 may affect various neurobiological activities such as sleeping, depression, and addiction. In this review, we focus on the neurobiological functions of Per2, which could help to better understand its roles in the CNS. Key Words: Circadian rhythm, Per2 gene, Sleep, Depression, Addiction, Neurotransmitter INTRODUCTION and lives in organisms because it can impart effects from the level of cells to organs including the brain. Thus, it is neces- A circadian rhythm is any physiological process that displays sary to understand clock-related genes that are controlling the a roughly 24 hour cycle in living beings, such as mammals, circadian rhythm endogenously. plants, fungi and cyanobacteria (Albrecht, 2012). In organ- The Period2 (Per2) gene is a member of the Period family isms, most biological functions such as sleeping and feeding of genes consisting of Per1, Per2, and Per3, and is mainly patterns are adapted to the circadian rhythm. -
Fosb Induction in Nucleus Accumbens by Cocaine Is Regulated by E2f3a
New Research Disorders of the Nervous System Fosb Induction in Nucleus Accumbens by Cocaine Is Regulated by E2F3a Hannah M. Cates,1 Casey K. Lardner,1 Rosemary C. Bagot,1 Rachael L. Neve,2 and Eric J. Nestler1 https://doi.org/10.1523/ENEURO.0325-18.2019 1Nash Family Department of Neuroscience and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029 and 2Gene Delivery Technology Core, Massachusetts General Hospital, Cambridge, Massachusetts 02139 Abstract The transcription factor ⌬FosB has been proposed as a molecular switch for the transition from casual, volitional drug use into a chronically addicted state, but the upstream regulatory mechanisms governing ⌬FosB expression are incompletely understood. In this study, we find a novel regulatory role for the transcription factor E2F3, recently implicated in transcriptional regulation by cocaine, in controlling ⌬FosB induction in the mouse nucleus accumbens (NAc) following cocaine administration. We find that an E2F consensus sequence 500 bp upstream of the Fosb transcription start site is enriched for E2F3 specifically over other E2F isoforms. We further conclude that ⌬FosB expression is regulated specifically by E2F3a, not E2F3b, that E2f3a expression is specific to D1 receptor-expressing medium spiny neurons, and that E2F3a overexpression in NAc recapitulates the induction of Fosb and ⌬Fosb mRNA expression observed after chronic cocaine exposure. E2F3a knockdown in NAc does not abolish ⌬Fosb induction by cocaine, a result consistent with previously published data showing that singular knockdown of upstream regulators of ⌬FosB is insufficient to block cocaine-induced expression. Finally, to elucidate potential combinatorial epigenetic mechanisms involved in E2F3a’s regulation of Fosb, we explore H3K4me3 enrichment at the Fosb promoter and find that it is not enhanced by E2F3a overexpression, suggesting that it may instead be a pre-existing permissive mark allowing for E2F3a to interact with Fosb. -

AP-1 in Cell Proliferation and Survival
Oncogene (2001) 20, 2390 ± 2400 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc AP-1 in cell proliferation and survival Eitan Shaulian1 and Michael Karin*,1 1Laboratory of Gene Regulation and Signal Transduction, Department of Pharmacology, University of California San Diego, 9500 Gilman Drive, La Jolla, California, CA 92093-0636, USA A plethora of physiological and pathological stimuli extensively discussed previously (Angel and Karin, induce and activate a group of DNA binding proteins 1991; Karin, 1995). that form AP-1 dimers. These proteins include the Jun, The mammalian AP-1 proteins are homodimers and Fos and ATF subgroups of transcription factors. Recent heterodimers composed of basic region-leucine zipper studies using cells and mice de®cient in individual AP-1 (bZIP) proteins that belong to the Jun (c-Jun, JunB proteins have begun to shed light on their physiological and JunD), Fos (c-Fos, FosB, Fra-1 and Fra-2), Jun functions in the control of cell proliferation, neoplastic dimerization partners (JDP1 and JDP2) and the closely transformation and apoptosis. Above all such studies related activating transcription factors (ATF2, LRF1/ have identi®ed some of the target genes that mediate the ATF3 and B-ATF) subfamilies (reviewed by (Angel eects of AP-1 proteins on cell proliferation and death. and Karin, 1991; Aronheim et al., 1997; Karin et al., There is evidence that AP-1 proteins, mostly those that 1997; Liebermann et al., 1998; Wisdom, 1999). In belong to the Jun group, control cell life and death addition, some of the Maf proteins (v-Maf, c-Maf and through their ability to regulate the expression and Nrl) can heterodimerize with c-Jun or c-Fos (Nishiza- function of cell cycle regulators such as Cyclin D1, p53, wa et al., 1989; Swaroop et al., 1992), whereas other p21cip1/waf1, p19ARF and p16. -
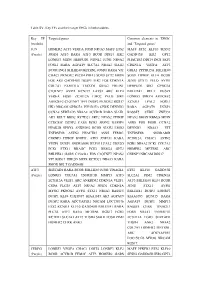
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

Epigenetic Regulation of Circadian Clocks and Its Involvement in Drug Addiction
G C A T T A C G G C A T genes Review Epigenetic Regulation of Circadian Clocks and Its Involvement in Drug Addiction Lamis Saad 1,2,3, Jean Zwiller 1,4, Andries Kalsbeek 2,3 and Patrick Anglard 1,5,* 1 Laboratoire de Neurosciences Cognitives et Adaptatives (LNCA), UMR 7364 CNRS, Université de Strasbourg, Neuropôle de Strasbourg, 67000 Strasbourg, France; [email protected] (L.S.); [email protected] (J.Z.) 2 The Netherlands Institute for Neuroscience (NIN), Royal Netherlands Academy of Arts and Sciences (KNAW), 1105 BA Amsterdam, The Netherlands; [email protected] 3 Department of Endocrinology and Metabolism, Amsterdam University Medical Center, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands 4 Centre National de la Recherche Scientifique (CNRS), 75016 Paris, France 5 Institut National de la Santé et de la Recherche Médicale (INSERM), 75013 Paris, France * Correspondence: [email protected]; Tel.: +33-03-6885-2009 Abstract: Based on studies describing an increased prevalence of addictive behaviours in several rare sleep disorders and shift workers, a relationship between circadian rhythms and addiction has been hinted for more than a decade. Although circadian rhythm alterations and molecular mechanisms associated with neuropsychiatric conditions are an area of active investigation, success is limited so far, and further investigations are required. Thus, even though compelling evidence connects the circadian clock to addictive behaviour and vice-versa, yet the functional mechanism behind this interaction remains largely unknown. At the molecular level, multiple mechanisms have been proposed to link the circadian timing system to addiction. The molecular mechanism of the circadian Citation: Saad, L.; Zwiller, J.; clock consists of a transcriptional/translational feedback system, with several regulatory loops, that Kalsbeek, A.; Anglard, P. -

T Suppressor Cells + CD8 Essential for the Differentiation of Human Micrornas by Ig-Like Transcript 3 Is Downregulation of Infla
Downregulation of Inflammatory MicroRNAs by Ig-like Transcript 3 Is Essential for the Differentiation of Human CD8 + T Suppressor Cells This information is current as of September 27, 2021. Chih-Chao Chang, Qing-Yin Zhang, Zhuoru Liu, Raphael A. Clynes, Nicole Suciu-Foca and George Vlad J Immunol 2012; 188:3042-3052; Prepublished online 2 March 2012; doi: 10.4049/jimmunol.1102899 Downloaded from http://www.jimmunol.org/content/188/7/3042 Supplementary http://www.jimmunol.org/content/suppl/2012/03/02/jimmunol.110289 http://www.jimmunol.org/ Material 9.DC1 References This article cites 46 articles, 20 of which you can access for free at: http://www.jimmunol.org/content/188/7/3042.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Downregulation of Inflammatory MicroRNAs by Ig-like Transcript 3 Is Essential for the Differentiation of Human CD8+ T Suppressor Cells Chih-Chao Chang, Qing-Yin Zhang, Zhuoru Liu, Raphael A. -

Differential Requirements for Tcf1 Long Isoforms in CD8+ and CD4+ T Cell Responses to Acute Viral Infection
Differential Requirements for Tcf1 Long Isoforms in CD8 + and CD4+ T Cell Responses to Acute Viral Infection This information is current as Jodi A. Gullicksrud, Fengyin Li, Shaojun Xing, Zhouhao of September 26, 2021. Zeng, Weiqun Peng, Vladimir P. Badovinac, John T. Harty and Hai-Hui Xue J Immunol 2017; 199:911-919; Prepublished online 26 June 2017; doi: 10.4049/jimmunol.1700595 Downloaded from http://www.jimmunol.org/content/199/3/911 Supplementary http://www.jimmunol.org/content/suppl/2017/06/24/jimmunol.170059 Material 5.DCSupplemental http://www.jimmunol.org/ References This article cites 40 articles, 11 of which you can access for free at: http://www.jimmunol.org/content/199/3/911.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 26, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Differential Requirements for Tcf1 Long Isoforms in CD8+ and CD4+ T Cell Responses to Acute Viral Infection Jodi A. -

Circadian- and Sex-Dependent Increases in Intravenous Cocaine Self-Administration in Npas2 Mutant Mice
Research Articles: Behavioral/Cognitive Circadian- and sex-dependent increases in intravenous cocaine self-administration in Npas2 mutant mice https://doi.org/10.1523/JNEUROSCI.1830-20.2020 Cite as: J. Neurosci 2020; 10.1523/JNEUROSCI.1830-20.2020 Received: 15 July 2020 Revised: 13 November 2020 Accepted: 18 November 2020 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2020 the authors 1 Circadian- and sex-dependent increases in intravenous cocaine self-administration in Npas2 mutant mice 2 3 Abbreviated title: Increased cocaine intake in female Npas2 mutants 4 5 Lauren M. DePoy1,2, Darius D. Becker-Krail1,2, Wei Zong3, Kaitlyn Petersen1,2, Neha M. Shah1, Jessica H. 6 Brandon1, Alyssa M. Miguelino1, George C. Tseng3, Ryan W. Logan1,2,4, *Colleen A. McClung1,2,4 7 8 1Department of Psychiatry, Translational Neuroscience Program, University of Pittsburgh School of Medicine, 9 15219 10 2Center for Neuroscience, University of Pittsburgh, 15261 11 3Department of Biostatistics, University of Pittsburgh, 15261 12 4Center for Systems Neurogenetics of Addiction, The Jackson Laboratory, 04609 13 14 15 *Corresponding Author: 16 Colleen A. McClung, PhD 17 450 Technology Dr. Ste 223 18 Pittsburgh, PA 15219 19 412-624-5547 20 [email protected] 21 22 Key words: circadian, sex-differences, cocaine, self-administration, substance use, Npas2 23 24 Pages: 37 25 Figures: 9 26 Extended data tables: 3 27 Abstract: 250 28 Introduction: 552 29 Discussion: 1490 30 31 Disclosures: 32 All authors have no financial disclosures or conflicts of interest to report. -

The Multifaceted Output of C-Jun Biological Activity: Focus at the Junction of CD8 T Cell Activation and Exhaustion
cells Review The Multifaceted Output of c-Jun Biological Activity: Focus at the Junction of CD8 T Cell Activation and Exhaustion Athanasios G. Papavassiliou 1 and Anna Maria Musti 2,* 1 Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, 11527 Athens, Greece; [email protected] 2 Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende, Italy * Correspondence: [email protected]; Tel.: +39-3337543732 Received: 22 September 2020; Accepted: 11 November 2020; Published: 13 November 2020 Abstract: c-Jun is a major component of the dimeric transcription factor activator protein-1 (AP-1), a paradigm for transcriptional response to extracellular signaling, whose components are basic-Leucine Zipper (bZIP) transcription factors of the Jun, Fos, activating transcription factor (ATF), ATF-like (BATF) and Jun dimerization protein 2 (JDP2) gene families. Extracellular signals regulate c-Jun/AP-1 activity at multiple levels, including transcriptional and posttranscriptional regulation of c-Jun expression and transactivity, in turn, establishing the magnitude and the duration of c-Jun/AP-1 activation. Another important level of c-Jun/AP-1 regulation is due to the capability of Jun family members to bind DNA as a heterodimer with every other member of the AP-1 family, and to interact with other classes of transcription factors, thereby acquiring the potential to integrate diverse extrinsic and intrinsic signals into combinatorial regulation of gene expression. Here, we review how these features of c-Jun/AP-1 regulation underlie the multifaceted output of c-Jun biological activity, eliciting quite distinct cellular responses, such as neoplastic transformation, differentiation and apoptosis, in different cell types.