Teleostei: Siluriformes: Callichthyidae) Com Base Em Seqüências De DNA Nuclear E Mitocondrial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

§4-71-6.5 LIST of CONDITIONALLY APPROVED ANIMALS November
§4-71-6.5 LIST OF CONDITIONALLY APPROVED ANIMALS November 28, 2006 SCIENTIFIC NAME COMMON NAME INVERTEBRATES PHYLUM Annelida CLASS Oligochaeta ORDER Plesiopora FAMILY Tubificidae Tubifex (all species in genus) worm, tubifex PHYLUM Arthropoda CLASS Crustacea ORDER Anostraca FAMILY Artemiidae Artemia (all species in genus) shrimp, brine ORDER Cladocera FAMILY Daphnidae Daphnia (all species in genus) flea, water ORDER Decapoda FAMILY Atelecyclidae Erimacrus isenbeckii crab, horsehair FAMILY Cancridae Cancer antennarius crab, California rock Cancer anthonyi crab, yellowstone Cancer borealis crab, Jonah Cancer magister crab, dungeness Cancer productus crab, rock (red) FAMILY Geryonidae Geryon affinis crab, golden FAMILY Lithodidae Paralithodes camtschatica crab, Alaskan king FAMILY Majidae Chionocetes bairdi crab, snow Chionocetes opilio crab, snow 1 CONDITIONAL ANIMAL LIST §4-71-6.5 SCIENTIFIC NAME COMMON NAME Chionocetes tanneri crab, snow FAMILY Nephropidae Homarus (all species in genus) lobster, true FAMILY Palaemonidae Macrobrachium lar shrimp, freshwater Macrobrachium rosenbergi prawn, giant long-legged FAMILY Palinuridae Jasus (all species in genus) crayfish, saltwater; lobster Panulirus argus lobster, Atlantic spiny Panulirus longipes femoristriga crayfish, saltwater Panulirus pencillatus lobster, spiny FAMILY Portunidae Callinectes sapidus crab, blue Scylla serrata crab, Samoan; serrate, swimming FAMILY Raninidae Ranina ranina crab, spanner; red frog, Hawaiian CLASS Insecta ORDER Coleoptera FAMILY Tenebrionidae Tenebrio molitor mealworm, -

Fauna Atingida Por Acidentes Ambientais Envolvendo Produtos Químicos
Universidade de São Paulo Escola Superior de Agricultura Luiz de Queiroz Departamento de Ciências do Solo Curso de Especialização em Gerenciamento Ambiental Sérgio Greif FAUNA ATINGIDA POR ACIDENTES AMBIENTAIS ENVOLVENDO PRODUTOS QUÍMICOS Orientadora: Biól. Iris Regina Fernandes Poffo (PhD.) São Paulo 2017 Universidade de São Paulo Escola Superior de Agricultura Luiz de Queiroz Departamento de Ciências do Solo Curso de Especialização em Gerenciamento Ambiental Sérgio Greif FAUNA ATINGIDA POR ACIDENTES AMBIENTAIS ENVOLVENDO PRODUTOS QUÍMICOS Orientadora: Biól. Iris Regina Fernandes Poffo (PhD.) Trabalho apresentado como pré-requisito para a obtenção de Certificado de Conclusão de Curso de Especialização em Gerenciamento Ambiental São Paulo 2017 iii “Nós nos tornamos, pelo poder de um glorioso acidente evolucionário chamado inteligência, mordomos da continuidade da vida na Terra. Não pedimos este papel, mas não podemos renegá-lo. Podemos não ser adequados para isso, mas aqui estamos." — Stephen Jay Gould iv SUMÁRIO SUMÁRIO......................................................................................................... iv . ......................................................................................... DEDICATÓRIA................................................................................................. vi ... ......................................................................................... AGRADECIMENTOS....................................................................................... vii . RELAÇÃO DE -

Cascadu, Flat-Head Or Chato
UWI The Online Guide to the Animals of Trinidad and Tobago Behaviour Callichthys callichthys (Flat-head Cascadu or Chato) Family: Callichthyidae (Plated Catfish) Order: Siluriformes (Catfish) Class: Actinopterygii (Ray-finned Fish) Fig. 1. Flat-head cascade, Callichthys callichthys. [“http://nas.er.usgs.gov/queries/factsheet.aspx?SpeciesID=335”, Downloaded 10th October 2011] TRAITS. Was first described by Linnaeus in 1758 and was named Silurus callichthys. They generally reach a maximum length of 20 cm and can weigh up to 80g.(Froese and Pauly, 2001) The females are generally larger and more robust as compared to the males. The Callichthys callichthys is an elongated catfish with a straight or flattened belly profile. It also has a broad flattened head and a body which is almost uniform in breath with some posterior tapering which beings after dorsal fin. (Figure 2) Its body consists of 2 rows of overlapping plates or scutes. Approximately 26 – 29 scutes seen on the upper lateral series and 25 – 28 scutes seen on the lower lateral series. The fins are rounded and the fish also has a total of 6-8 soft dorsal rays. It also has 2 pairs of maxillary barbles near its mouth and small eyes. The fish has an inferior type mouth ( Berra, 2007). The Callichthys callichthys is dark olive green in colour to a grey brown as seen in Figure 1, with the males having a blue to violet sheen on its flanks. ECOLOGY. Callichthys callichthys is a freshwater organism which is primarily riverine in habitat (Arratia, 2003). Only 2 families of catfishes are found to colonise marine habitats UWI The Online Guide to the Animals of Trinidad and Tobago Behaviour (Arratia, 2003). -

Siluriformes: Callichthyidae) in the Subterranean Domain of Northern and Northeastern Brazil
13 4 297 Tencatt et al NOTES ON GEOGRAPHIC DISTRIBUTION Check List 13 (4): 297–303 https://doi.org/10.15560/13.4.297 First report of armored catfishes Callichthyinae Bonaparte, 1838 (Siluriformes: Callichthyidae) in the subterranean domain of northern and northeastern Brazil Luiz Fernando Caserta Tencatt,1 Bruno Ferreira dos Santos,2 Maria Elina Bichuette3 1 Universidade Estadual de Maringá, Coleção Ictiológica do Núcleo de Pesquisas em Limnologia, Ictiologia e Aquicultura e Programa de Pós- Graduação em Ecologia de Ambientes Aquáticos Continentais, Av. Colombo, 5790, 87020-900 Maringá, Paraná, Brazil. 2 Universidade Federal de Mato Grosso do Sul, Programa de Pós-Graduação em Biologia Animal, Av. Costa e Silva, 79070-900 Campo Grande, Mato Grosso do Sul, Brazil. 3 Universidade Federal de São Carlos, Departamento de Ecologia e Biologia Evolutiva, Laboratório de Estudos Subterrâneos, Rodovia Washington Luis, km 235, 13565-905 São Carlos, São Paulo, Brazil. Corresponding author: Luiz Fernando Caserta Tencatt, [email protected] Abstract The first occurrence of the armored catfishes of the subfamily Callichthynae is reported in subterranean water bodies of northern and northeastern Brazil. The records include 3 species, each occurring in 1 of the 3 caves in the central and northeastern regions of Brazil: Callichthys callichthys from Casa do Caboclo cave, Sergipe state; Hoplosternum lit- torale from the Gruna da Lagoa do Meio, Bahia state; and Megalechis thoracata, from Casa de Pedra cave, Tocantins state. Keywords Camboatá, cave, hypogean habitat, karstic areas, Neotropical region. Academic editor: Bárbara Calegari | Received 2 March 2017 | Accepted 10 June 2017 | Published 14 August 2017 Citation: Tencatt LFC, Ferreira dos Santos B, Bichuette ME (2017) First report of armored catfishes Callichthyinae( Bonaparte, 1838) (Siluriformes: Callichthyidae) in the subterranean domain. -

Documento Completo Descargar Archivo
Publicaciones científicas del Dr. Raúl A. Ringuelet Zoogeografía y ecología de los peces de aguas continentales de la Argentina y consideraciones sobre las áreas ictiológicas de América del Sur Ecosur, 2(3): 1-122, 1975 Contribución Científica N° 52 al Instituto de Limnología Versión electrónica por: Catalina Julia Saravia (CIC) Instituto de Limnología “Dr. Raúl A. Ringuelet” Enero de 2004 1 Zoogeografía y ecología de los peces de aguas continentales de la Argentina y consideraciones sobre las áreas ictiológicas de América del Sur RAÚL A. RINGUELET SUMMARY: The zoogeography and ecology of fresh water fishes from Argentina and comments on ichthyogeography of South America. This study comprises a critical review of relevant literature on the fish fauna, genocentres, means of dispersal, barriers, ecological groups, coactions, and ecological causality of distribution, including an analysis of allotopic species in the lame lake or pond, the application of indexes of diversity of severa¡ biotopes and comments on historical factors. Its wide scope allows to clarify several aspects of South American Ichthyogeography. The location of Argentina ichthyological fauna according to the above mentioned distributional scheme as well as its relation with the most important hydrography systems are also provided, followed by additional information on its distribution in the Argentine Republic, including an analysis through the application of Simpson's similitude test in several localities. SINOPSIS I. Introducción II. Las hipótesis paleogeográficas de Hermann von Ihering III. La ictiogeografía de Carl H. Eigenmann IV. Estudios de Emiliano J. Mac Donagh sobre distribución de peces argentinos de agua dulce V. El esquema de Pozzi según el patrón hidrográfico actual VI. -

Summary Report of Freshwater Nonindigenous Aquatic Species in U.S
Summary Report of Freshwater Nonindigenous Aquatic Species in U.S. Fish and Wildlife Service Region 4—An Update April 2013 Prepared by: Pam L. Fuller, Amy J. Benson, and Matthew J. Cannister U.S. Geological Survey Southeast Ecological Science Center Gainesville, Florida Prepared for: U.S. Fish and Wildlife Service Southeast Region Atlanta, Georgia Cover Photos: Silver Carp, Hypophthalmichthys molitrix – Auburn University Giant Applesnail, Pomacea maculata – David Knott Straightedge Crayfish, Procambarus hayi – U.S. Forest Service i Table of Contents Table of Contents ...................................................................................................................................... ii List of Figures ............................................................................................................................................ v List of Tables ............................................................................................................................................ vi INTRODUCTION ............................................................................................................................................. 1 Overview of Region 4 Introductions Since 2000 ....................................................................................... 1 Format of Species Accounts ...................................................................................................................... 2 Explanation of Maps ................................................................................................................................ -

Global Catfish Biodiversity 17
American Fisheries Society Symposium 77:15–37, 2011 © 2011 by the American Fisheries Society Global Catfi sh Biodiversity JONATHAN W. ARMBRUSTER* Department of Biological Sciences, Auburn University 331 Funchess, Auburn University, Alabama 36849, USA Abstract.—Catfi shes are a broadly distributed order of freshwater fi shes with 3,407 cur- rently valid species. In this paper, I review the different clades of catfi shes, all catfi sh fami- lies, and provide information on some of the more interesting aspects of catfi sh biology that express the great diversity that is present in the order. I also discuss the results of the widely successful All Catfi sh Species Inventory Project. Introduction proximately 10.8% of all fi shes and 5.5% of all ver- tebrates are catfi shes. Renowned herpetologist and ecologist Archie Carr’s But would every one be able to identify the 1941 parody of dichotomous keys, A Subjective Key loricariid catfi sh Pseudancistrus pectegenitor as a to the Fishes of Alachua County, Florida, begins catfi sh (Figure 2A)? It does not have scales, but it with “Any damn fool knows a catfi sh.” Carr is right does have bony plates. It is very fl at, and its mouth but only in part. Catfi shes (the Siluriformes) occur has long jaws but could not be called large. There is on every continent (even fossils are known from a barbel, but you might not recognize it as one as it Antarctica; Figure 1); and the order is extremely is just a small extension of the lip. There are spines well supported by numerous complex synapomor- at the front of the dorsal and pectoral fi ns, but they phies (shared, derived characteristics; Fink and are not sharp like in the typical catfi sh. -

BMC Evolutionary Biology, 2014, 14
Abe et al. BMC Evolutionary Biology 2014, 14:152 http://www.biomedcentral.com/1471-2148/14/152 RESEARCH ARTICLE Open Access Systematic and historical biogeography of the Bryconidae (Ostariophysi: Characiformes) suggesting a new rearrangement of its genera and an old origin of Mesoamerican ichthyofauna Kelly T Abe, Tatiane C Mariguela, Gleisy S Avelino, Fausto Foresti and Claudio Oliveira* Abstract Background: Recent molecular hypotheses suggest that some traditional suprageneric taxa of Characiformes require revision, as they may not constitute monophyletic groups. This is the case for the Bryconidae. Various studies have proposed that this family (considered a subfamily by some authors) may be composed of different genera. However, until now, no phylogenetic study of all putative genera has been conducted. Results: In the present study, we analyzed 27 species (46 specimens) of all currently recognized genera of the Bryconidae (ingroup) and 208 species representing all other families and most genera of the Characiformes (outgroup). Five genes were sequenced: 16SrRNA, Cytochrome b, recombination activating gene 1 and 2 and myosin heavy chain 6 cardiac muscle. The final matrix contained 4699 bp and was analyzed by maximum likelihood, maximum parsimony and Bayesian analyses. The results show that the Bryconidae, composed of Brycon, Chilobrycon, Henochilus and Salminus, is monophyletic and is the sister group of Gasteropelecidae + Triportheidae. However, the genus Brycon is polyphyletic. Fossil studies suggest that the family originated approximately 47 million years ago (Ma) and that one of the two main lineages persisted only in trans-Andean rivers, including Central American rivers, suggesting a much older origin of Mesoamerican ichthyofauna than previously accepted. -
Stream Ichthyofauna of the Tapajós National Forest, Pará, Brazil
A peer-reviewed open-access journal ZooKeys 580: 125–144 (2016)Stream ichthyofauna of the Tapajós National Forest, Pará, Brazil 125 doi: 10.3897/zookeys.580.6659 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research Stream ichthyofauna of the Tapajós National Forest, Pará, Brazil Cárlison Silva-Oliveira1, André Luiz Colares Canto2, Frank Raynner Vasconcelos Ribeiro1.2 1 Programa de Pós-Graduação em Recursos Aquáticos Continentais Amazônicos, Instituto de Ciências e Tecno- logia das Águas, Universidade Federal do Oeste do Pará. Rua Vera Paz, s/n 68035-110 Santarém, Pará, Brazil 2 Universidade Federal do Oeste do Pará, Curso de Bacharelado em Ciências Biológicas. Campus Amazônia, sala 232. Avenida Mendonça Furtado, 2946, 68040-470 Santarém, Pará, Brazil Corresponding author: Cárlison Silva-Oliveira ([email protected]) Academic editor: N. Bogutskaya | Received 23 September 2015 | Accepted 7 March 2016 | Published 12 April 2016 http://zoobank.org/D03C4745-036A-4ED9-8BA3-C8D58F9563D9 Citation: Silva-Oliveira C, Canto ALC, Ribeiro FRV (2016) Stream ichthyofauna of the Tapajós National Forest, Pará, Brazil. ZooKeys 580: 125–144. doi: 10.3897/zookeys.580.6659 Abstract The fish fauna of freshwater streams in the Tapajos National Forest was surveyed and a list of species is presented. The sampling was conducted from 2012 to 2013 during the dry season. Fish were collected with dip nets and seine nets in 22 streams of 1st to 3rd order. Sampling resulted in 3035 specimens belong- ing to 117 species, 27 families and six orders. The most abundant species were Bryconops aff. melanurus, Hemigrammus belottii, and Hemigrammus analis. Four undescribed species were recognized, one of which is known only from the area of this study. -
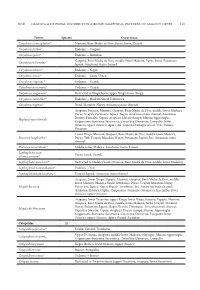
DAGOSTA & DE PINNA: DISTRIBUTION & BIOGEOGRAPHICAL PATTERNS of AMAZON FISHES Taxon Species Occurrence Corydoras Stenocep
2019 DAGOSTA & DE PINNA: DISTRIBUTION & BIOGEOGRAPHICAL PATTERNS OF AMAZON FISHES 113 Taxon Species Occurrence Corydoras stenocephalus* Mamoré, Beni-Madre de Dios, Purus, Juruá, Ucayali Corydoras sterbai* Endemic – Guaporé Corydoras sychri* Endemic – Marañon Guaporé, Beni-Madre de Dios, middle-lower Madeira, Purus, Juruá, Putumayo, Corydoras trilineatus* Japurá, Amazonas main channel Corydoras tukano* Endemic – Negro Corydoras urucu* Endemic – Coari-Urucu Corydoras virginiae* Endemic – Ucayali Corydoras weitzmani* Endemic – Ucayali Corydoras xinguensis* Restricted to Xingu basin (upper Xingu, lower Xingu) Corydoras zawadzkii* Endemic – Madeira Shield Tributaries Corydoras zygatus* Juruá, Marañon-Nanay, Amazonas main channel Araguaia, Juruena, Mamoré, Guaporé, Beni-Madre de Dios, middle-lower Madeira, Purus, Ucayali, Putumayo, Japurá, Negro, Amazonas main channel, Amazonas Estuary, Parnaíba, Capim, Araguari-Macari-Amapá, Maroni-Approuague, Hoplosternum littorale Coppename-Suriname-Saramacca, Corentyne-Demerara, Essequibo, lower Orinoco, upper Orinoco, Apure, Atl. Coastal Drainages of Col. Ven., Paraná- Paraguay Lower Xingu, Mamoré, Guaporé, Beni-Madre de Dios, middle-lower Madeira, Dianema longibarbis* Purus, Tefé, Ucayali, Marañon-Nanay, Putumayo, Japurá, Jari, Amazonas main channel Dianema urostriatum* Middle-lower Madeira, Amazonas main channel Lepthoplosternum Purus, Juruá, Ucayali altamazonicum* Lepthoplosternum beni* Restricted to Madeira basin (Mamoré, Beni-Madre de Dios, middle-lower Madeira) Lepthoplosternum stellatum* Endemic -

Mineral Accumulation in the Catfish Megalechis Personata
The Journal of Experimental Biology 202, 2121–2129 (1999) 2121 Printed in Great Britain © The Company of Biologists Limited 1999 JEB1967 EFFECT OF LOW AMBIENT MINERAL CONCENTRATIONS ON THE ACCUMULATION OF CALCIUM, MAGNESIUM AND PHOSPHORUS BY EARLY LIFE STAGES OF THE AIR-BREATHING ARMOURED CATFISH MEGALECHIS PERSONATA (SILURIFORMES: CALLICHTHYIDAE) JAN H. MOL1,*, WIM ATSMA2, GERT FLIK2, HANS BOUWMEESTER3 AND JAN W. M. OSSE3 1University of Suriname, CELOS, PO Box 9212, Paramaribo, Surinam, 2Department of Animal Physiology, University of Nijmegen, Toernooiveld 25, 6525 ED Nijmegen, The Netherlands and 3Department of Experimental Animal Morphology and Cell Biology, Wageningen Agricultural University, PO Box 338, 6700 AH Wageningen, The Netherlands *e-mail: [email protected] Accepted 24 April; published on WWW 7 July 1999 Summary The accumulation of calcium, magnesium and respiration in the third week after hatching did not affect phosphorus was measured during an 8-week period in the rates of mineral accumulation. The high rates of early life stages of the air-breathing armoured catfish accumulation of calcium and magnesium of M. personata Megalechis personata acclimated to low-mineral fresh in LMF of 654 and 58 µmol h−1 kg−1, respectively, exceed water (0.073 mmol l−1 calcium, 0.015 mmol l−1 magnesium, the rates of uptake of calcium and magnesium of teleosts <0.001 mmol l−1 phosphate) and high-mineral fresh water reported in the literature. The high rates of mineral (0.59 mmol l−1 calcium, 1.94 mmol l−1 magnesium, accumulation in the early life stages of M. personata reflect <0.001 mmol l−1 phosphate). -

Pre-Hispanic Fishing Practices in Interfluvial
Pre-Hispanic fishing practices in interfluvial Amazonia: Zooarchaeological evidence from managed landscapes on the Llanos de Mojos savanna Gabriela Prestes-Carneiro, Philippe Béarez, Myrtle Shock, Heiko Prümers, Carla Jaimes Betancourt To cite this version: Gabriela Prestes-Carneiro, Philippe Béarez, Myrtle Shock, Heiko Prümers, Carla Jaimes Betan- court. Pre-Hispanic fishing practices in interfluvial Amazonia: Zooarchaeological evidence from man- aged landscapes on the Llanos de Mojos savanna. PLoS ONE, Public Library of Science, 2019, 10.1371/journal.pone.0214638. hal-02401142 HAL Id: hal-02401142 https://hal.archives-ouvertes.fr/hal-02401142 Submitted on 9 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. RESEARCH ARTICLE Pre-Hispanic fishing practices in interfluvial Amazonia: Zooarchaeological evidence from managed landscapes on the Llanos de Mojos savanna Gabriela Prestes-Carneiro1,2*, Philippe BeÂarez2, Myrtle Pearl Shock1, Heiko PruÈ mers3, Carla Jaimes Betancourt4 a1111111111 1 Anthropology and Archaeology Program, Institute of Social