Supplementary Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alembic Pot Still
ALEMBIC POT STILL INSTRUCTION MANUAL CAN BE USED WITH THE GRAINFATHER OR T500 BOILER SAFETY Warning: This system produces a highly flammable liquid. PRECAUTION: • Always use the Alembic Pot Still System in a room with adequate ventilation. • Never leave the Alembic Pot Still system unattended when operating. • Keep the Alembic Pot Still system away from all sources of ignition, including smoking, sparks, heat, and open flames. • Ensure all other equipment near to the Alembic Pot Still system or the alcohol is earthed. • A fire extinguishing media suitable for alcohol should be kept nearby. This can be water fog, fine water spray, foam, dry powder, carbon dioxide, sand or dolomite. • Do not boil dry. In the event the still is boiled dry, reset the cutout button under the base of the still. In the very unlikely event this cutout fails, a fusible link gives an added protection. IN CASE OF SPILLAGE: • Shut off all possible sources of ignition. • Clean up spills immediately using cloth, paper towels or other absorbent materials such as soil, sand or other inert material. • Collect, seal and dispose accordingly • Mop area with excess water. CONTENTS Important points before getting started ............................................................................... 3 Preparing the Alembic Pot Still ................................................................................................. 5 Distilling a Whiskey, Rum or Brandy .......................................................................................7 Distilling neutral -

Review of Extractive Distillation. Process Design, Operation
Review of Extractive Distillation. Process design, operation optimization and control Vincent Gerbaud, Ivonne Rodríguez-Donis, Laszlo Hegely, Péter Láng, Ferenc Dénes, Xinqiang You To cite this version: Vincent Gerbaud, Ivonne Rodríguez-Donis, Laszlo Hegely, Péter Láng, Ferenc Dénes, et al.. Review of Extractive Distillation. Process design, operation optimization and control. Chemical Engineering Research and Design, Elsevier, 2019, 141, pp.229-271. 10.1016/j.cherd.2018.09.020. hal-02161920 HAL Id: hal-02161920 https://hal.archives-ouvertes.fr/hal-02161920 Submitted on 21 Jun 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Open Archive Toulouse Archive Ouverte OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible This is an author’s version published in: http://oatao.univ-toulouse.fr/239894 Official URL: https://doi.org/10.1016/j.cherd.2018.09.020 To cite this version: Gerbaud, Vincent and Rodríguez-Donis, Ivonne and Hegely, Laszlo and Láng, Péter and Dénes, Ferenc and You, Xinqiang Review of Extractive Distillation. Process design, operation optimization and control. (2018) Chemical Engineering Research and Design, 141. -

Minimum Reflux in Liquid–Liquid Extraction
17th European Symposium on Computer Aided Process Engineering – ESCAPE17 V. Plesu and P.S. Agachi (Editors) © 2007 Elsevier B.V. All rights reserved. 1 Minimum Reflux in Liquid–Liquid Extraction Santanu Bandyopadhyaya and Calin-Cristian Cormosb aEnergy Systems Engineering, Indian Institute of Technology, Bombay, Powai, Mumbai 400 076, India, E-mail: [email protected] bDepartment of Chemical Engineering, Faculty of Chemistry and Chemical Engineering, University "Babeş-Bolyai", Arany Janos 11, Cluj-Napoca 400028, Romania, E-mail: [email protected] Abstract In a simple countercurrent arrangement of different stages of liquid–liquid extraction operation, the richest extract leaving the operation is in equilibrium with the feed. However, by using reflux it is possible to enrich the extract further. A simple counter current liquid–liquid extraction operation with reflux is analogous in its essentials to distillation. In this paper, the method based on Invariant Rectifying-Stripping (IRS) curves, originally proposed to calculate minimum reflux and minimum energy requirement in distillation, has been extended to liquid–liquid extraction. The equivalent IRS curves for a ternary liquid–liquid extraction predicts the feed location in the counter current process. It also predicts the minimum reflux requirement for a given separation and minimum amount of solvent required. Keywords: liquid extraction, IRS curves, feed location, minimum solvent. 1. Introduction Liquid–liquid extraction is a separation process that takes advantage of the distribution of a substance between two insoluble liquids phases [1]. Feed to be separated is mixed with extracting solvent to produce a solvent-rich phase, called extract, and a solvent-lean phase, called raffinate. -

Reflux Condensation in Narrow Rectangular Channels with Perforated Fins Nadia Souidi, André Bontemps
Reflux condensation in narrow rectangular channels with perforated fins Nadia Souidi, André Bontemps To cite this version: Nadia Souidi, André Bontemps. Reflux condensation in narrow rectangular channels with perforated fins. Applied Thermal Engineering, Elsevier, 2003, 23, pp.871-891. 10.1016/S1359-4311(03)00021-8. hal-00184135 HAL Id: hal-00184135 https://hal.archives-ouvertes.fr/hal-00184135 Submitted on 19 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Reflux condensation in narrow rectangular channels with perforated fins N. Souidi a, A. Bontemps b,* a GRETh-CEA Grenoble, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France b LEGI-GRETh, Universitee Joseph Fourier, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France Reflux condensation is an industrial process that aims to reduce the content of the less volatile com- ponent or to eliminate the non-condensable phase of a vapour mixture, by the means of separation. Separation consists in condensing the less volatile phase and to recover the condensate while simulta- neously, the non-condensable species are recuperated at the top of the system. Compact plate-fin heat exchangers can be used in gas separation processes. -

United States Patent (10) Patent No.: US 9,586,922 B2 Wood Et Al
USOO9586922B2 (12) United States Patent (10) Patent No.: US 9,586,922 B2 Wood et al. (45) Date of Patent: Mar. 7, 2017 (54) METHODS FOR PURIFYING 9,388,150 B2 * 7/2016 Kim ..................... CO7D 307/48 5-(HALOMETHYL)FURFURAL 9,388,151 B2 7/2016 Browning et al. 2007/O161795 A1 7/2007 Cvak et al. 2009, 0234142 A1 9, 2009 Mascal (71) Applicant: MICROMIDAS, INC., West 2010, 0083565 A1 4, 2010 Gruter Sacramento, CA (US) 2010/0210745 A1 8/2010 McDaniel et al. 2011 O144359 A1 6, 2011 Heide et al. (72) Inventors: Alex B. Wood, Sacramento, CA (US); 2014/01 00378 A1 4/2014 Masuno et al. Shawn M. Browning, Sacramento, CA 2014/O1878O2 A1 7/2014 Mikochik et al. (US);US): Makoto N. Masuno,M. s Elk Grove, s 2015/02668432015,0203462 A1 9/20157/2015 CahanaBrowning et etal. al. CA (US); Ryan L. Smith, Sacramento, 2016/0002190 A1 1/2016 Browning et al. CA (US); John Bissell, II, Sacramento, 2016,01681 07 A1 6/2016 Masuno et al. CA (US) 2016/02O7897 A1 7, 2016 Wood et al. (73) Assignee: MICROMIDAS, INC., West FOREIGN PATENT DOCUMENTS Sacramento, CA (US) CN 101475544. A T 2009 (*) Notice: Subject to any disclaimer, the term of this N 1939: A 658 patent is extended or adjusted under 35 DE 635,783 C 9, 1936 U.S.C. 154(b) by 0 days. EP 291494 A2 11, 1988 EP 1049657 B1 3, 2003 (21) Appl. No.: 14/852,306 GB 1220851 A 1, 1971 GB 1448489 A 9, 1976 RU 2429234 C2 9, 2011 (22) Filed: Sep. -

Heterogeneous Azeotropic Distillation
PROSIMPLUS APPLICATION EXAMPLE HETEROGENEOUS AZEOTROPIC DISTILLATION EXAMPLE PURPOSE This example illustrates a high purity separation process of an azeotropic mixture (ethanol-water) through heterogeneous azeotropic distillation. This process includes distillation columns. Additionally these rigorous multi- stage separation modules are part of a recycling stream, demonstrating the efficiency of ProSimPlus convergence methods. Specifications are set on output streams in order to insure the required purity. This example illustrates the way to set "non-standard" specifications in the multi-stage separation modules. Three phase calculations (vapor-liquid- liquid) are done with the taken into account of possible liquid phase splitting. ACCESS Free-Internet Restricted to ProSim clients Restricted Confidential CORRESPONDING PROSIMPLUS FILE PSPS_EX_EN-Heterogeneous-Azeotropic-Distillation.pmp3 . Reader is reminded that this use case is only an example and should not be used for other purposes. Although this example is based on actual case it may not be considered as typical nor are the data used always the most accurate available. ProSim shall have no responsibility or liability for damages arising out of or related to the use of the results of calculations based on this example. Copyright © 2009 ProSim, Labège, France - All rights reserved www.prosim.net Heterogeneous azeotropic distillation Version: March, 2009 Page: 2 / 12 TABLE OF CONTENTS 1. PROCESS MODELING 3 1.1. Process description 3 1.2. Process flowsheet 5 1.3. Specifications 6 1.4. Components 6 1.5. Thermodynamic model 7 1.6. Operating conditions 7 1.7. "Hints and Tips" 9 2. RESULTS 9 2.1. Comments on results 9 2.2. Mass and energy balances 10 2.3. -

Distillation Accessscience from McgrawHill Education
6/19/2017 Distillation AccessScience from McGrawHill Education (http://www.accessscience.com/) Distillation Article by: King, C. Judson University of California, Berkeley, California. Last updated: 2014 DOI: https://doi.org/10.1036/10978542.201100 (https://doi.org/10.1036/10978542.201100) Content Hide Simple distillations Fractional distillation Vaporliquid equilibria Distillation pressure Molecular distillation Extractive and azeotropic distillation Enhancing energy efficiency Computational methods Stage efficiency Links to Primary Literature Additional Readings A method for separating homogeneous mixtures based upon equilibration of liquid and vapor phases. Substances that differ in volatility appear in different proportions in vapor and liquid phases at equilibrium with one another. Thus, vaporizing part of a volatile liquid produces vapor and liquid products that differ in composition. This outcome constitutes a separation among the components in the original liquid. Through appropriate configurations of repeated vaporliquid contactings, the degree of separation among components differing in volatility can be increased manyfold. See also: Phase equilibrium (/content/phaseequilibrium/505500) Distillation is by far the most common method of separation in the petroleum, natural gas, and petrochemical industries. Its many applications in other industries include air fractionation, solvent recovery and recycling, separation of light isotopes such as hydrogen and deuterium, and production of alcoholic beverages, flavors, fatty acids, and food oils. Simple distillations The two most elementary forms of distillation are a continuous equilibrium distillation and a simple batch distillation (Fig. 1). http://www.accessscience.com/content/distillation/201100 1/10 6/19/2017 Distillation AccessScience from McGrawHill Education Fig. 1 Simple distillations. (a) Continuous equilibrium distillation. -

Building a Home Distillation Apparatus
BUILDING A HOME DISTILLATION APPARATUS A Step by Step Guide Building a Home Distillation Apparatus i BUILDING A HOME DISTILLATION APPARATUS Foreword The pages that follow contain a step-by-step guide to building a relatively sophisticated distillation apparatus from commonly available materials, using simple tools, and at a cost of under $100 USD. The information contained on this site is directed at anyone who may want to know more about the subject: students, hobbyists, tinkers, pure water enthusiasts, survivors, the curious, and perhaps even amateur wine and beer makers. Designing and building this apparatus is the only subject of this manual. You will find that it confines itself solely to those areas. It does not enter into the domains of fermentation, recipes for making mash, beer, wine or any other spirits. These areas are covered in detail in other readily available books and numerous web sites. The site contains two separate design plans for the stills. And while both can be used for a number of distillation tasks, it should be recognized that their designs have been optimized for the task of separating ethyl alcohol from a water-based mixture. Having said that, remember that the real purpose of this site is to educate and inform those of you who are interested in this subject. It is not to be construed in any fashion as an encouragement to break the law. If you believe the law is incorrect, please take the time to contact your representatives in government, cast your vote at the polls, write newsletters to the media, and in general, try to make the changes in a legal and democratic manner. -

Batch Distillation of Spirits: Experimental Study and Simulation
Research article Received: 5 April 2018 Revised: 11 January 2019 Accepted: 15 January 2019 Published online in Wiley Online Library (wileyonlinelibrary.com) DOI 10.1002/jib.560 Batch distillation of spirits: experimental study and simulation of the behaviour of volatile aroma compounds Adrien Douady,1 Cristian Puentes,1 Pierre Awad1,2 and Martine Esteban-Decloux1* This paper focuses on the behaviour of volatile compounds during batch distillation of wine or low wine, in traditional Charentais copper stills, heated with a direct open flame at laboratory (600 L) and industrial (2500 L) scale. Sixty-nine volatile compounds plus ethanol were analysed during the low wine distillation in the 600 L alembic still. Forty-four were quantified and classified according to their concentration profile in the distillate over time and compared with previous studies. Based on the online re- cording of volume flow, density and temperature of the distillate with a Coriolis flowmeter, distillation was simulated with ProSim® BatchColumn software. Twenty-six volatile compounds were taken into account, using the coefficients of the ‘Non- Random Two Liquids’ model. The concentration profiles of 18 compounds were accurately represented, with slight differences in the maximum concentration for seven species together with a single compound that was poorly represented. The distribution of the volatile compounds in the four distillate fractions (heads, heart, seconds and tails) was well estimated by simulation. Fi- nally, data from wine and low wine distillations in the large-scale alembic still (2500 L) were correctly simulated, suggesting that it was possible to adjust the simulation parameters with the Coriolis flowmeter recording and represent the concentration pro- files of most of the quantifiable volatile compounds. -

Tissue Free Water Tritium Separation from Foodstuffs by Azeotropic Distillation
140 SK98K0361 21st RHDJasna podChopkom TISSUE FREE WATER TRITIUM SEPARATION FROM FOODSTUFFS BY AZEOTROPIC DISTILLATION Florentina Constantin, Ariadna Ciubotaru, DecebalPopa Inspectorate of Public Health of Bucharest, Romania 1. INTRODUCTION The actual tritium content in the environment is due to both natural production by cosmic ray interactions in the upper atmosphere and to varying amount of man- made tritium. Tritium, the radioactive isotope of hydrogen, is one of the most important radionuclide released to the environment during the normal operation of a typical nuclear power reactor. Tritiated water constitutes almost the entire radioactivity in liquid waste discharged to the environment from heavy water reactors. Therefore, monitoring tritium in air, precipitation, drinking water and vegetation is necessary for assessing the environmental impact of tritium releases in gaseous and liquid effluents from nuclear plants. The use for agricultural purposes of river water receiving releases of tritium, results in contamination of irrigated crops and then in the contamination of the food chain. ? Tritium is everywhere in living matter, has a long physical half-time (T= 12.26 y) and is known to follow protium pathways in biological material. Tritium in liquid or solid biota samples can not be easily determined quantitatively by any kind of nondestructive analysis because its beta radiation energy (E =18 keV) is so weak that the most part is absorbed in the sample. Therefore, most non-aqueous samples must be treated chemically or physically to be suitable for counting. The choice of preparation method will depend on the type of samples, number and quantities of samples to be processed and availability of measurement equipment. -
Minimum Reflux Ratio
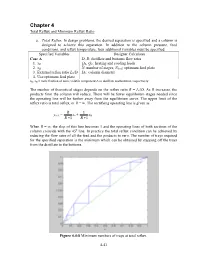
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux. -

Comparison of Complete Extractive and Azeotropic Distillation Processes for Anhydrous Ethanol Production Using Aspen Plustm Simu
43 A publication of CHEMICAL ENGINEERING TRANSACTIONS VOL. 80, 2020 The Italian Association of Chemical Engineering Online at www.cetjournal.it Guest Editors: Eliseo Maria Ranzi, Rubens Maciel Filho Copyright © 2020, AIDIC Servizi S.r.l. DOI: 10.3303/CET2080008 ISBN 978-88-95608-78-5; ISSN 2283-9216 Comparison of Complete Extractive and Azeotropic Distillation Processes for Anhydrous Ethanol Production using Aspen PlusTM Simulator Nahieh T. Miranda*, Rubens Maciel Filho, Maria Regina Wolf Maciel Laboratory of Optimization, Design, and Advanced Control (LOPCA)/Laboratory of Development of Separation Processes (LDPS), School of Chemical Engineering, University of Campinas, Brazil. [email protected] Performance of the extractive and azeotropic distillation processes using ethylene glycol and cyclohexane as solvents, respectively, for anhydrous ethanol production, were investigated using 02 RadFrac columns each and solvent recycling streams via simulation in Aspen Plus™ Simulator. Both operate at 1 atm, and feed flow -1 rate equals to 100 kmol.h (ethanol – 0.896 and water – 0.104 in mole fractions at the azeotropic point). The NRTL-RK was the model used for extractive and azeotropic distillations. The anhydrous ethanol purity from top of the 1st column of the extractive distillation (22 stages) was 99.50 % (on a mole basis) and water with nd st 99.20 % of purity at the top of the 2 column. On the other hand, in the azeotropic distillation, the 1 column (30 stages) had a bottom product of anhydrous ethanol purity of 99.99 % and water with 99.99 % of purity in the 2nd column, also at the bottom. Both processes produced anhydrous ethanol with a high-grade purity required by the standard norms ASTM D4806, EN 15376, and ANP 36.