Can Weighting Improve Bushy Trees? Models of Cytochrome B Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Point Reyes National Seashore Bird List
Birds of Point Reyes National Seashore Gaviidae (Loons) Alcedinidae (Kingfishers) Podicipedidae (Grebes) Picidae (Woodpeckers) Diomedeidae (Albatrosses) Tyrannidae (Tyrant Flycatcher) Procellariidae (Shearwaters, Petrels) Alaudidae (Larks) Hydrobatidae (Storm Petrels) Hirundinidae (Swallows) Sulidae (Boobies, Gannets) Laniidae (Shrikes) Pelecanidae (Pelicans) Vireonidae (Vireos) Phalacrocoracidae (Cormorants) Corvidae (Crows, Jays) Fregatidae (Frigate Birds) Paridae (Chickadees, Titmice) Ardeidae (Herons, Bitterns, & Egrets) Aegithalidae (Bushtits) Threskiornithidae (Ibises, Spoonbills) Sittidae (Nuthatches) Ciconiidae (Storks) Certhiidae (Creepers) Anatidae (Ducks, Geese, Swans) Troglodytidae (Wrens) Cathartidae (New World Vultures) Cinclidae (Dippers) Accipitridae (Hawks, Kites, Eagles) & Regulidae (Kinglets) Falconidae (Caracaras, Falcons) Sylviidae (Old World Warblers, Gnatcatchers) Odontophoridae (New World Quail) Turdidae (Thrushes) Rallidae (Rails, Gallinules, Coots) Timaliidae (Babblers) Gruidae (Cranes) Mimidae (Mockingbirds, Thrashers) Charadriidae (Lapwings, Plovers) Motacillidae (Wagtails, Pipits) Haematopodidae (Oystercatcher) Bombycillidae (Waxwings) Recurvirostridae (Stilts, Avocets) Ptilogonatidae (Silky-flycatcher) Scolopacidae (Sandpipers, Phalaropes) Parulidae (Wood Warblers) Laridae (Skuas, Gulls, Terns, Skimmers) Cardinalidae (Cardinals) Alcidae (Auks, Murres, Puffins) Emberizidae (Emberizids) Columbidae (Pigeons, Doves) Fringillidae (Finches) Cuculidae (Cuckoos, Road Runners, Anis) NON-NATIVES Tytonidae (Barn Owls) -
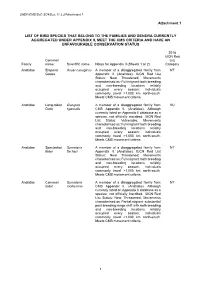
Attachment 1 LIST of BIRD SPECIES THAT BELONG to the FAMILIES
UNEP/CMS/ScC-SC4/Doc.11.3.2/Attachment 1 Attachment 1 LIST OF BIRD SPECIES THAT BELONG TO THE FAMILIES AND GENERA CURRENTLY AGGREGATED UNDER APPENDIX II, MEET THE CMS CRITERIA AND HAVE AN UNFAVOURABLE CONSERVATION STATUS 2018 IUCN Red Common List Family name Scientific name Notes for Appendix II (Sheets 1 or 2) Category Anatidae Emperor Anser canagicus A member of a disaggregated family from NT Goose Appendix II. (Anatidae). IUCN Red List Status: Near Threatened; Movements characterised as: Full migrant: both breeding and non-breeding locations reliably occupied every season; individuals commonly travel >1,000 km north-south. Meets CMS movement criteria. Anatidae Long-tailed Clangula A member of a disaggregated family from VU Duck hyemalis CMS Appendix II. (Anatidae). Although currently listed on Appendix II database as a species, not officially inscribed. IUCN Red List Status: Vulnerable; Movements characterised as: Full migrant: both breeding and non-breeding locations reliably occupied every season; individuals commonly travel >1,000 km north-south. Meets CMS movement criteria. Anatidae Spectacled Somateria A member of a disaggregated family from NT Eider fischeri Appendix II. (Anatidae). IUCN Red List Status: Near Threatened; Movements characterised as: Full migrant: both breeding and non-breeding locations reliably occupied every season; individuals commonly travel >1,000 km north-south. Meets CMS movement criteria. Anatidae Common Somateria A member of a disaggregated family from NT Eider mollissima CMS Appendix II. (Anatidae). Although currently listed on Appendix II database as a species, not officially inscribed. IUCN Red List Status: Near Threatened; Movements characterised as: Partial migrant: substantial post-breeding range shift with both breeding and non-breeding locations reliably occupied every season; individuals commonly travel >1,000 km north-south. -

Predicting the Distribution of the Vulnerable Yellow-Breasted Pipit (Anthus Chloris) Using Species Distribution Modelling
1 Predicting the distribution of the Vulnerable Yellow-breasted Pipit (Anthus chloris) using Species Distribution Modelling Darren W. Pietersen1*, Ian T. Little2, Raymond Jansen3 and Andrew E. McKechnie1 1DST-NRF Centre of Excellence at the Percy FitzPatrick Institute, Department of Zoology and Entomology, University of Pretoria, Private Bag X20, Hatfield, 0028, South Africa 2The Endangered Wildlife Trust, Private Bag X11, Modderfontein, 1645, Johannesburg, South Africa 3Department of Environmental, Water and Earth Sciences, Tshwane University of Technology, Private Bag X680, Pretoria, 0001, South Africa *Address for correspondence: Department of Zoology and Entomology University of Pretoria Private Bag X20 Hatfield 0028 [email protected] +27 82 937 6052 2 Abstract The Yellow-breasted Pipit (Anthus chloris) is endemic to the eastern escarpment of South Africa, marginally entering eastern Lesotho. This species is classified as globally Vulnerable due to a perceived decreasing population size and loss of habitat. We employed Species Distribution Modelling to investigate the predicted range of this species to determine whether additional purportedly suitable habitat exists where this species may be found, and to assess the degree to which habitat loss has affected this species. We used a database of 250 independently obtained and verified sightings to predict the summer breeding distribution of this species and compare our verified sightings and predicted range to the sightings currently in the Second Southern African Bird Atlas Project (SABAP2) database and the latest regional conservation assessment. Our models closely approximate the current distribution of the Yellow-breasted Pipit, and suggest that most of the purportedly suitable habitat is occupied, at least at the macro-scale. -

South Puget Sound Streaked Horned Lark (Eremophila Alpestris Strigata) Genetic Rescue Study Report for Year 2
South Puget Sound Streaked Horned Lark (Eremophila alpestris strigata) Genetic Rescue Study Report for Year 2 Spring/Summer 2012 Photo credit: Rod Gilbert South Puget Sound Streaked Horned Lark (Eremophila alpestris strigata) Genetic Rescue Study Draft Report for Year 2 September 2012 Prepared by Adrian Wolf THE CENTER FOR NATURAL LANDS MANAGEMENT The Center for Natural Lands Management 120 East Union Avenue, suite 215 Olympia, WA 98501 Tel. 360-742-8212 Email: [email protected] Abstract Hatchability of Streaked Horned Lark (Eremophila alpestris strigata) eggs in the Puget Lowlands of Washington State is extremely low relative to other grassland nesting birds at the same site and generally. Because genetic factors (inbreeding depression) appear to be a likely explanation, an effort to increase genetic diversity was initiated in 2011. 2012 was the second year of the genetic rescue effort initiated at the 13th Division Prairie on Joint Base Lewis-McChord. A total of eight breeding lark pairs and one unpaired male were detected within the study area. Nest building was first detected on 25 April; the first eggs were observed on 16 May; the first nestlings hatched on or around 27 May, the first fledgling was observed on 20 June. Twenty nests were located, which produced a total of 49 eggs (2.9 eggs/nest ± 0.9 SD). Mean number of eggs per completed clutch (n = 15) was 3.1 ± 0.6 (SD), an increase from 2.3 in 2011. A total of 14 local nestlings were color-banded, and at least four of the fourteen were observed foraging independently. Hatchability of the Puget Sound nests increased to 84% in 2012, from 61% in 2011. -

Field Checklist (PDF)
Surf Scoter Marbled Godwit OWLS (Strigidae) Common Raven White-winged Scoter Ruddy Turnstone Eastern Screech Owl CHICKADEES (Paridae) Common Goldeneye Red Knot Great Horned Owl Black-capped Chickadee Barrow’s Goldeneye Sanderling Snowy Owl Boreal Chickadee Bufflehead Semipalmated Sandpiper Northern Hawk-Owl Tufted Titmouse Hooded Merganser Western Sandpiper Barred Owl NUTHATCHES (Sittidae) Common Merganser Least Sandpiper Great Gray Owl Red-breasted Nuthatch Red-breasted Merganser White-rumped Sandpiper Long-eared Owl White-breasted Nuthatch Ruddy Duck Baird’s Sandpiper Short-eared Owl CREEPERS (Certhiidae) VULTURES (Cathartidae) Pectoral Sandpiper Northern Saw-Whet Owl Brown Creeper Turkey Vulture Purple Sandpiper NIGHTJARS (Caprimulgidae) WRENS (Troglodytidae) HAWKS & EAGLES (Accipitridae) Dunlin Common Nighthawk Carolina Wren Osprey Stilt Sandpiper Whip-poor-will House Wren Bald Eagle Buff-breasted Sandpiper SWIFTS (Apodidae) Winter Wren Northern Harrier Ruff Chimney Swift Marsh Wren Sharp-shinned Hawk Short-billed Dowitcher HUMMINGBIRDS (Trochilidae) THRUSHES (Muscicapidae) Cooper’s Hawk Wilson’s Snipe Ruby-throated Hummingbird Golden-crowned Kinglet Northern Goshawk American Woodcock KINGFISHERS (Alcedinidae) Ruby-crowned Kinglet Red-shouldered Hawk Wilson’s Phalarope Belted Kingfisher Blue-gray Gnatcatcher Broad-winged Hawk Red-necked Phalarope WOODPECKERS (Picidae) Eastern Bluebird Red-tailed Hawk Red Phalarope Red-headed Woodpecker Veery Rough-legged Hawk GULLS & TERNS (Laridae) Yellow-bellied Sapsucker Gray-cheeked Thrush Golden -

Bontebok Birds
Birds recorded in the Bontebok National Park 8 Little Grebe 446 European Roller 55 White-breasted Cormorant 451 African Hoopoe 58 Reed Cormorant 465 Acacia Pied Barbet 60 African Darter 469 Red-fronted Tinkerbird * 62 Grey Heron 474 Greater Honeyguide 63 Black-headed Heron 476 Lesser Honeyguide 65 Purple Heron 480 Ground Woodpecker 66 Great Egret 486 Cardinal Woodpecker 68 Yellow-billed Egret 488 Olive Woodpecker 71 Cattle Egret 494 Rufous-naped Lark * 81 Hamerkop 495 Cape Clapper Lark 83 White Stork n/a Agulhas Longbilled Lark 84 Black Stork 502 Karoo Lark 91 African Sacred Ibis 504 Red Lark * 94 Hadeda Ibis 506 Spike-heeled Lark 95 African Spoonbill 507 Red-capped Lark 102 Egyptian Goose 512 Thick-billed Lark 103 South African Shelduck 518 Barn Swallow 104 Yellow-billed Duck 520 White-throated Swallow 105 African Black Duck 523 Pearl-breasted Swallow 106 Cape Teal 526 Greater Striped Swallow 108 Red-billed Teal 529 Rock Martin 112 Cape Shoveler 530 Common House-Martin 113 Southern Pochard 533 Brown-throated Martin 116 Spur-winged Goose 534 Banded Martin 118 Secretarybird 536 Black Sawwing 122 Cape Vulture 541 Fork-tailed Drongo 126 Black (Yellow-billed) Kite 547 Cape Crow 127 Black-shouldered Kite 548 Pied Crow 131 Verreauxs' Eagle 550 White-necked Raven 136 Booted Eagle 551 Grey Tit 140 Martial Eagle 557 Cape Penduline-Tit 148 African Fish-Eagle 566 Cape Bulbul 149 Steppe Buzzard 572 Sombre Greenbul 152 Jackal Buzzard 577 Olive Thrush 155 Rufous-chested Sparrowhawk 582 Sentinel Rock-Thrush 158 Black Sparrowhawk 587 Capped Wheatear -

Birds of Gus Engeling Wildlife Management Area
TEXAS PARKS AND WILDLIFE BIRDS OF G U S E N G E L I N G WILDLIFE MANAGEMENT AREA A FIELD CHECKLIST “Act Natural” Visit a Wildlife Management Area at our Web site: http://www.tpwd.state.tx.us Cover: Illustration of Pileated Woodpecker by Rob Fleming. HABITAT DESCRIPTION he Gus Engeling Wildlife Management Area is located in the northwest corner of Anderson County, 20 miles Tnorthwest of Palestine, Texas, on U.S. Highway 287. The management area contains 10,958 acres of land owned by the Texas Parks and Wildlife Department. Most of the land was purchased in 1950 and 1951, with the addition of several smaller tracts through 1960. It was originally called the Derden Wildlife Management Area, but was later changed to the Engeling Wildlife Management Area in honor of Biologist Gus A. Engeling, who was killed by a poacher on the area in December 1951. The area is drained by Catfish Creek which is a tributary of the Trinity River. The topography is gently rolling to hilly, with a well-defined drainage system that empties into Catfish Creek. Most of the small streams are spring fed and normally flow year-round. The soils are mostly light colored, rapidly permeable sands on the upland, and moderately permeable, gray-brown, sandy loams in the bottomland along Catfish Creek. The climate is classified as moist, sub-humid, with an annual rainfall of about 40 inches. The vegetation consists of deciduous forest with an overstory made up of oak, hickory, sweetgum and elm; with associated understory species of dogwood, American beautyberry, huckleberry, greenbrier, etc. -

The Evolutionary History of the White Wagtail Species Complex, (Passeriformes: Motacillidae: Motacilla Alba)
Contributions to Zoology 88 (2019) 257-276 CTOZ brill.com/ctoz The evolutionary history of the white wagtail species complex, (Passeriformes: Motacillidae: Motacilla alba) Maliheh Pirayesh Shirazinejad Department of Biology, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran Mansour Aliabadian Department of Biology, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran Research Department of Zoological Innovations, Institute of Applied Zoology, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran [email protected] Omid Mirshamsi Department of Biology, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran Research Department of Zoological Innovations, Institute of Applied Zoology, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran Abstract The white wagtail (Motacilla alba) species complex with its distinctive plumage in separate geographical areas can serve as a model to test evolutionary hypotheses. Its extensive variety in plumage, despite the genetic similarity between taxa, and the evolutionary events connected to this variety are poorly under- stood. Therefore we sampled in the breeding range of the white wagtail: 338 individuals were analyzed from 74 areas in the Palearctic and Mediterranean. We studied the white wagtail complex based on two mitochondrial DNA markers to make inferences about the evolutionary history. Our phylogenetic trees highlight mtDNA sequences (ND2, CR), and one nuclear marker (CHD1Z), which partly correspond to earlier described clades: the northern Palearctic (clade N); eastern and central Asia (clade SE); south- western Asia west to the British Isles (clade SW); and Morocco (clade M). The divergence of all clades occurred during the Pleistocene. We also used ecological niche modelling for three genetic lineages (ex- cluding clade M); results showed congruence between niche and phylogenetic divergence in these clades. -

Interspecific Social Dominance Mimicry in Birds
bs_bs_banner Zoological Journal of the Linnean Society, 2014. With 6 figures Interspecific social dominance mimicry in birds RICHARD OWEN PRUM1,2* 1Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT 06520-8150, USA 2Peabody Natural History Museum, Yale University, New Haven, CT 06520-8150, USA Received 3 May 2014; revised 17 June 2014; accepted for publication 21 July 2014 Interspecific social dominance mimicry (ISDM) is a proposed form of social parasitism in which a subordinate species evolves to mimic and deceive a dominant ecological competitor in order to avoid attack by the dominant, model species. The evolutionary plausibility of ISDM has been established previously by the Hairy-Downy game (Prum & Samuelson). Psychophysical models of avian visual acuity support the plausibility of visual ISDM at distances ∼>2–3 m for non-raptorial birds, and ∼>20 m for raptors. Fifty phylogenetically independent examples of avian ISDM involving 60 model and 93 mimic species, subspecies, and morphs from 30 families are proposed and reviewed. Patterns of size differences, phylogeny, and coevolutionary radiation generally support the predic- tions of ISDM. Mimics average 56–58% of the body mass of the proposed model species. Mimics may achieve a large potential deceptive social advantage with <20% reduction in linear body size, which is well within the range of plausible, visual size confusion. Several, multispecies mimicry complexes are proposed (e.g. kiskadee- type flycatchers) which may coevolve through hierarchical variation in the deceptive benefits, similar to Müllerian mimicry. ISDM in birds should be tested further with phylogenetic, ecological, and experimental investigations of convergent similarity in appearance, ecological competition, and aggressive social interactions between sympatric species. -

South Africa Mega Birding III 5Th to 27Th October 2019 (23 Days) Trip Report
South Africa Mega Birding III 5th to 27th October 2019 (23 days) Trip Report The near-endemic Gorgeous Bushshrike by Daniel Keith Danckwerts Tour leader: Daniel Keith Danckwerts Trip Report – RBT South Africa – Mega Birding III 2019 2 Tour Summary South Africa supports the highest number of endemic species of any African country and is therefore of obvious appeal to birders. This South Africa mega tour covered virtually the entire country in little over a month – amounting to an estimated 10 000km – and targeted every single endemic and near-endemic species! We were successful in finding virtually all of the targets and some of our highlights included a pair of mythical Hottentot Buttonquails, the critically endangered Rudd’s Lark, both Cape, and Drakensburg Rockjumpers, Orange-breasted Sunbird, Pink-throated Twinspot, Southern Tchagra, the scarce Knysna Woodpecker, both Northern and Southern Black Korhaans, and Bush Blackcap. We additionally enjoyed better-than-ever sightings of the tricky Barratt’s Warbler, aptly named Gorgeous Bushshrike, Crested Guineafowl, and Eastern Nicator to just name a few. Any trip to South Africa would be incomplete without mammals and our tally of 60 species included such difficult animals as the Aardvark, Aardwolf, Southern African Hedgehog, Bat-eared Fox, Smith’s Red Rock Hare and both Sable and Roan Antelopes. This really was a trip like no other! ____________________________________________________________________________________ Tour in Detail Our first full day of the tour began with a short walk through the gardens of our quaint guesthouse in Johannesburg. Here we enjoyed sightings of the delightful Red-headed Finch, small numbers of Southern Red Bishops including several males that were busy moulting into their summer breeding plumage, the near-endemic Karoo Thrush, Cape White-eye, Grey-headed Gull, Hadada Ibis, Southern Masked Weaver, Speckled Mousebird, African Palm Swift and the Laughing, Ring-necked and Red-eyed Doves. -

White Wagtails Motacilla Alba in Victoria
VOL. 16 (1) MARCH 1995 21 AUSTRALIAN BIRD WATCHER 1995, 16, 21-33 White Wagtails Motacilla alba in Victoria by MIKE CARTER1, ROBERT FARNES2 and NEVILLE PAMMENP 130 Canadian Bay Road, Mt Eliza, Victoria 3930 2P.O. Box 475, Portland, Victoria 3305 3Department of Chemical Engineering, University of Melbourne, Parkville, Victoria 3052 Summary Two White' Wagtails Motacilla alba seen in Victoria in 1992 constitute the first fully authenticated records for the state and the third and fourth acceptable records for Australia. One was identified subspecifically as leucopsis and the other as baicalensis or an intergrade with that form, whereas a previous Western Australian record was ocularis. This is an account of these occurrences together with discussion of their likely provenance and the features used for identification to subspecific level, age and sex. It is suggested that one bird was moulting on a revised timetable now in phase with the austral rather than boreal seasons. Introduction Five species of wagtails (Motacillidae) have been recorded in Australia but only one is generally recognised as appearing regularly. The Yellow Wagtail Motacilla jlava is an annual non-breeding summer visitor which occurs most abundantly in the tropical wet areas of Western Australia and the Northern Territory, and more occasional! y in. Queensland. A second species, the Grey Wagtail M. cinerea, is probably also a regular but rare summer visitor to Arnhem Land, N.T. (Carter 1993a). Elsewhere both are only rare vagrants. The other species, Citrine (Yellow-headed) Wagtail M. citreola, White Wagtail M. alba and Black-backed Wagtail M. lugens, are very rare vagrants. -

Multi-Locus Phylogeny of African Pipits and Longclaws (Aves: Motacillidae) Highlights Taxonomic Inconsistencies
Running head: African pipit and longclaw taxonomy Multi-locus phylogeny of African pipits and longclaws (Aves: Motacillidae) highlights taxonomic inconsistencies DARREN W. PIETERSEN,1* ANDREW E. MCKECHNIE,1,2 RAYMOND JANSEN,3 IAN T. LITTLE4 AND ARMANDA D.S. BASTOS5 1DST-NRF Centre of Excellence at the Percy FitzPatrick Institute, Department of Zoology and Entomology, University of Pretoria, Hatfield, South Africa 2South African Research Chair in Conservation Physiology, National Zoological Garden, South African National Biodiversity Institute, P.O. Box 754, Pretoria 0001, South Africa 3Department of Environmental, Water and Earth Sciences, Tshwane University of Technology, Pretoria, South Africa 4Endangered Wildlife Trust, Johannesburg, South Africa 5Department of Zoology and Entomology, University of Pretoria, Hatfield, South Africa *Corresponding author. Email: [email protected] 1 Abstract The globally distributed avian family Motacillidae consists of 5–7 genera (Anthus, Dendronanthus, Tmetothylacus, Macronyx and Motacilla, and depending on the taxonomy followed, Amaurocichla and Madanga) and 66–68 recognised species, of which 32 species in four genera occur in sub- Saharan Africa. The taxonomy of the Motacillidae has been contentious, with variable numbers of genera, species and subspecies proposed and some studies suggesting greater taxonomic diversity than what is currently (five genera and 67 species) recognised. Using one nuclear (Mb) and two mitochondrial (cyt b and CO1) gene regions amplified from DNA extracted from contemporary and museum specimens, we investigated the taxonomic status of 56 of the currently recognised motacillid species and present the most taxonomically complete and expanded phylogeny of this family to date. Our results suggest that the family comprises six clades broadly reflecting continental distributions: sub-Saharan Africa (two clades), the New World (one clade), Palaearctic (one clade), a widespread large-bodied Anthus clade, and a sixth widespread genus, Motacilla.