Genomic Scans for Selective Sweeps Through Haplotype Homozygosity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony
Supplementary Data Th2 and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in UBIOPRED Chih-Hsi Scott Kuo1.2, Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony Rowe3, Iaonnis Pandis2, Ana Sousa4, Julie Corfield5, Ratko Djukanovic6, Rene 7 7 8 2 1† Lutter , Peter J. Sterk , Charles Auffray , Yike Guo , Ian M. Adcock & Kian Fan 1†* # Chung on behalf of the U-BIOPRED consortium project team 1Airways Disease, National Heart & Lung Institute, Imperial College London, & Biomedical Research Unit, Biomedical Research Unit, Royal Brompton & Harefield NHS Trust, London, United Kingdom; 2Department of Computing & Data Science Institute, Imperial College London, United Kingdom; 3Janssen Research and Development, High Wycombe, Buckinghamshire, United Kingdom; 4Respiratory Therapeutic Unit, GSK, Stockley Park, United Kingdom; 5AstraZeneca R&D Molndal, Sweden and Areteva R&D, Nottingham, United Kingdom; 6Faculty of Medicine, Southampton University, Southampton, United Kingdom; 7Faculty of Medicine, University of Amsterdam, Amsterdam, Netherlands; 8European Institute for Systems Biology and Medicine, CNRS-ENS-UCBL, Université de Lyon, France. †Contributed equally #Consortium project team members are listed under Supplementary 1 Materials *To whom correspondence should be addressed: [email protected] 2 List of the U-BIOPRED Consortium project team members Uruj Hoda & Christos Rossios, Airways Disease, National Heart & Lung Institute, Imperial College London, UK & Biomedical Research Unit, Biomedical Research Unit, Royal -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

CSE642 Final Version
Eindhoven University of Technology MASTER Dimensionality reduction of gene expression data Arts, S. Award date: 2018 Link to publication Disclaimer This document contains a student thesis (bachelor's or master's), as authored by a student at Eindhoven University of Technology. Student theses are made available in the TU/e repository upon obtaining the required degree. The grade received is not published on the document as presented in the repository. The required complexity or quality of research of student theses may vary by program, and the required minimum study period may vary in duration. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain Eindhoven University of Technology MASTER THESIS Dimensionality Reduction of Gene Expression Data Author: S. (Sako) Arts Daily Supervisor: dr. V. (Vlado) Menkovski Graduation Committee: dr. V. (Vlado) Menkovski dr. D.C. (Decebal) Mocanu dr. N. (Nikolay) Yakovets May 16, 2018 v1.0 Abstract The focus of this thesis is dimensionality reduction of gene expression data. I propose and test a framework that deploys linear prediction algorithms resulting in a reduced set of selected genes relevant to a specified case. Abstract In cancer research there is a large need to automate parts of the process of diagnosis, this is mainly to reduce cost, make it faster and more accurate. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Theranostics Deficiency of ATP6V1H Causes Bone Loss by Inhibiting
Theranostics 2016, Vol. 6, Issue 12 2183 Ivyspring International Publisher Theranostics 2016; 6(12): 2183-2195. doi: 10.7150/thno.17140 Research Paper Deficiency of ATP6V1H Causes Bone Loss by Inhibiting Bone Resorption and Bone Formation through the TGF-β1 Pathway Xiaohong Duan1*, Jin Liu1*, Xueni Zheng1*, Zhe Wang1, Yanli Zhang1, Ying Hao1, Tielin Yang2, Hongwen Deng3 1. State Key Laboratory of Military Stomatology, National Clinical Research Center for Oral Diseases, Shaanxi Key Laboratory of Oral Diseases, Department of Oral Biology, Clinic of Oral Rare and Genetic Diseases, School of Stomatology, The Fourth Military Medical University, Xi’an, 710032, People’s Republic of China. 2. Key Laboratory of Biomedical Information Engineering of Ministry of Education, and Institute of Molecular Genetics, School of Life Science and Technology, Xi’an Jiaotong University, Xi’an 710049, People’s Republic of China. 3. School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA 70112, USA. *Contributed equally to this work. Corresponding author: Xiaohong Duan, State Key Laboratory of Military Stomatology, National Clinical Research Center for Oral Diseases, Shaanxi Key Laboratory of Oral Diseases, Department of Oral Biology, Clinic of Oral Rare and Genetic Diseases, School of Stomatology, the Fourth Military Medical University, 145 West Changle Road, Xi’an 710032, P. R. China. Tel: 86-29-84776169; Fax: 86-29-84776169; E-mail: [email protected]. © Ivyspring International Publisher. Reproduction is permitted for personal, noncommercial use, provided that the article is in whole, unmodified, and properly cited. See http://ivyspring.com/terms for terms and conditions. Received: 2016.08.08; Accepted: 2016.08.15; Published: 2016.09.13 Abstract Vacuolar-type H +-ATPase (V-ATPase) is a highly conserved, ancient enzyme that couples the energy of ATP hydrolysis to proton transport across vesicular and plasma membranes of eukaryotic cells. -
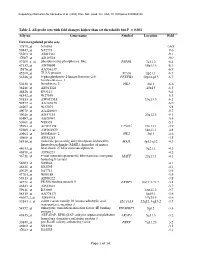
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Discovery of Molecular Mechanisms Underlying Lysosomal and Mitochondrial Defects in Parkinson’S Disease
Discovery of Molecular Mechanisms Underlying Lysosomal and Mitochondrial Defects In Parkinson’s Disease Brigitte Phillips Supervisor: Associate Professor Antony Cooper A thesis in fulfillment of the requirements for the degree of Doctor of Philosophy St Vincent’s Clinical School, Faculty of Medicine The University of New South Wales & The Garvan Institute of Medical Research April, 2018 THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Phillips First name: Brigitte Other names/s: Radinovic Abbreviation for degree as given in the University calendar: PhD School: St Vincent’s Clinical School Title: The emerging contributions of the lysosome and mitochondria to Parkinson’s diseases Abstract 350 words maximum Parkinson’s disease (PD) is a common, debilitating neurodegenerative disease yet the causes of cell dysfunction in PD remain unclear. By integrating available patient data with data from an unbiased assessment of proteomic changes in multiple cellular PD models, this study has identified new aspects of mitochondrial and lysosomal dysfunction that likely contribute to PD. Two areas were investigated in detail; the mitochondrial protein CHCHD2 and the V-ATPase complex, which acidifies endolysosomal compartments. CHCHD2 had not been well characterized or associated with PD when identified in this study, however PD-causative variants have since been described and CHCHD2 has recently been proposed to regulate mitochondrial cristae structure and interact with cytochrome c. Data from this thesis extends CHCHD2 dysfunction to sporadic PD patients, where its reduced expression was identified in the brain. In exploring the potential function of CHCHD2, mitochondrial impairment resulted in rapid translational up- regulation of CHCHD2 and its specific accumulation in depolarised mitochondria, suggesting that CHCHD2 plays a targeted role in aiding mitochondrial recovery or quarantining cytochrome c in response to mitochondrial damage. -

Kidney V-Atpase-Rich Cell Proteome Database
A comprehensive list of the proteins that are expressed in V-ATPase-rich cells harvested from the kidneys based on the isolation by enzymatic digestion and fluorescence-activated cell sorting (FACS) from transgenic B1-EGFP mice, which express EGFP under the control of the promoter of the V-ATPase-B1 subunit. In these mice, type A and B intercalated cells and connecting segment principal cells of the kidney express EGFP. The protein identification was performed by LC-MS/MS using an LTQ tandem mass spectrometer (Thermo Fisher Scientific). For questions or comments please contact Sylvie Breton ([email protected]) or Mark A. Knepper ([email protected]). -

Molecular Architecture Underlying Fluid Absorption by the Developing Inner
RESEARCH ARTICLE Molecular architecture underlying fluid absorption by the developing inner ear Keiji Honda1†, Sung Huhn Kim2‡, Michael C Kelly3, Joseph C Burns3§, Laura Constance2, Xiangming Li2#, Fei Zhou2, Michael Hoa4, Matthew W Kelley3, Philine Wangemann2*, Robert J Morell5, Andrew J Griffith1* 1Molecular Biology and Genetics Section, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, United States; 2Anatomy and Physiology Department, Kansas State University, Manhattan, United States; 3Developmental Neuroscience Section, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, United States; 4Auditory Development and Restoration Program, National Institute on *For correspondence: Deafness and Other Communication Disorders, National Institutes of Health, [email protected] (PW); Bethesda, United States; 5Genomics and Computational Biology Core, National [email protected] (AJG) Institute on Deafness and Other Communication Disorders, National Institutes of Present address: Health, Bethesda, United States †Otolaryngology Department, Tsuchiura Kyodo General Hospital, Tsuchiura, Japan; ‡Department of Abstract Mutations of SLC26A4 are a common cause of hearing loss associated with Otorhinolaryngology, Head and enlargement of the endolymphatic sac (EES). Slc26a4 expression in the developing mouse Neck Surgery, Yonsei University College of Medicine, Seoul, endolymphatic sac is required for acquisition of normal inner ear structure and function. Here, we Korea; §Decibel Therapeutics, show that the mouse endolymphatic sac absorbs fluid in an SLC26A4-dependent fashion. Fluid Cambridge, United States; absorption was sensitive to ouabain and gadolinium but insensitive to benzamil, bafilomycin and #Technique R and D-Drug S3226. Single-cell RNA-seq analysis of pre- and postnatal endolymphatic sacs demonstrates two Substance, GlaxoSmithKline types of differentiated cells. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2. -

Caractérisation De Nouveaux Gènes Et Polymorphismes Potentiellement Impliqués Dans Les Interactions Hôtes-Pathogènes
Aix-Marseille Université, Faculté de Médecine de Marseille Ecole Doctorale des Sciences de la Vie et de la Santé THÈSE DE DOCTORAT Présentée par Charbel ABOU-KHATER Date et lieu de naissance: 08-Juilllet-1990, Zahlé, LIBAN En vue de l’obtention du grade de Docteur de l’Université d’Aix-Marseille Mention: Biologie, Spécialité: Microbiologie Caractérisation de nouveaux gènes et polymorphismes potentiellement impliqués dans les interactions hôtes-pathogènes Publiquement soutenue le 5 Juillet 2017 devant le jury composé de : Pr. Daniel OLIVE Directeur de Thèse Pr. Brigitte CROUAU-ROY Rapporteur Dr. Benoît FAVIER Rapporteur Dr. Pierre PONTAROTTI Examinateur Thèse codirigée par Pr. Daniel OLIVE et Dr Laurent ABI-RACHED Laboratoires d’accueil URMITE Research Unit on Emerging Infectious and Tropical Diseases, UMR 6236, Faculty of Medicine, 27, Boulevard Jean Moulin, 13385 Marseille, France CRCM, Centre de Recherche en Cancérologie de Marseille,Inserm 1068, 27 Boulevard Leï Roure, BP 30059, 13273 Marseille Cedex 09, France 2 Acknowledgements First and foremost, praises and thanks to God, Holy Mighty, Holy Immortal, All-Holy Trinity, for His showers of blessings throughout my whole life and to whom I owe my very existence. Glory to the Father, and to the Son, and to the Holy Spirit: now and ever and unto ages of ages. I would like to express my sincere gratitude to my advisors Prof. Daniel Olive and Dr. Laurent Abi-Rached, for the continuous support, for their patience, motivation, and immense knowledge. Someday, I hope to be just like you. A special thanks to my “Godfather” who perfectly fulfilled his role, Dr. -

Figure S1. 17-Mer Distribution in the Yangtze Finless Porpoise Genome
Figure S1. 17-mer distribution in the Yangtze finless porpoise genome. The x-axis is 17-mer depth (X); the y-axis is the number of sequencing reads at that depth. Figure S2. Sequence depth distribution of the assembly data. The x-axis shows the sequencing depth (X) and the y-axis shows the number of bases at a given depth. The results demonstrate that 99% of bases sequencing depth is more than 20. Figure S3. Comparison of gene structure characteristics of Yangtze finless porpoise and other cetaceans. The x-axis represents the length of corresponding genetic element of exon number and the y-axis represents gene density. Figure S4. Phylogeny relationships between the Yangtze finless porpoise and other mammals reconstructed by RAxML with the GTR+G+I model. Table S1. Summary of sequenced reads Raw Reads Qualified Reads1 Total Read Sequence Physical Total Read Sequence Physical Library SRA Data Length Coverage2 Coverage2 Data Length Coverage2 Coverage2 Insert Size (bp) Number (Gb) (bp) (×) (×) (Gb) (bp) (×) (×) 289 58.94 150.00 23.67 22.80 57.84 149.75 23.23 22.41 SRR6923836 462 71.33 150.00 28.65 44.12 70.12 149.74 28.16 43.44 SRR6923837 624 67.47 150.00 27.10 56.36 63.90 149.67 25.66 53.50 SRR6923834 791 57.58 150.00 23.12 60.97 55.39 149.67 22.24 58.78 SRR6923835 4,000 108.73 150.00 43.67 582.22 70.74 150.00 28.41 378.80 SRR6923832 7,000 115.4 150.00 46.35 1,081.39 84.76 150.00 34.04 794.27 SRR6923833 11,000 107.37 150.00 43.12 1,581.08 79.78 150.00 32.04 1,174.81 SRR6923830 18,000 127.46 150.00 51.19 3,071.33 97.75 150.00 39.26 2,355.42 SRR6923831 Total 714.28 - 286.87 6,500.27 580.28 - 233.04 4,881.43 - 1Raw reads in mate-paired libraries were filtered to remove duplicates and reads with low quality and/or adapter contamination, raw reads in paired-end libraries were filtered in the same manner then subjected to k-mer-based correction.