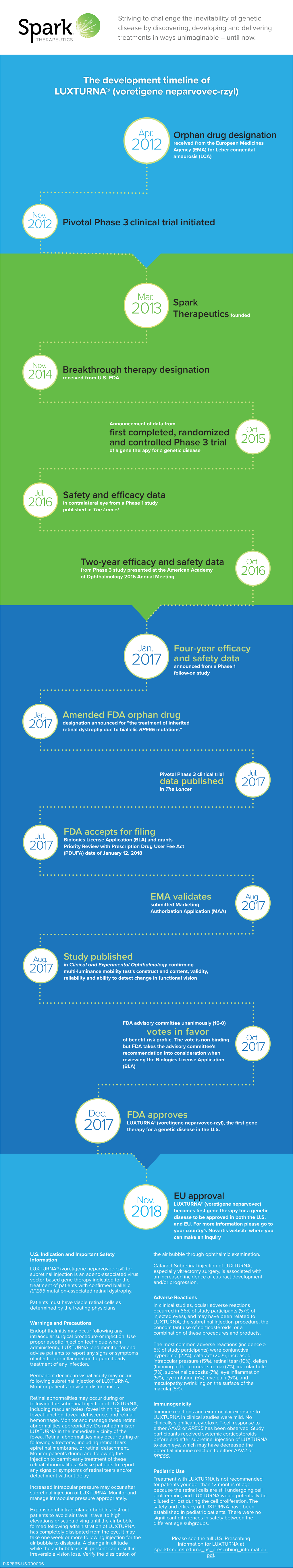
The Development Timeline of LUXTURNA® (Voretigene Neparvovec-Rzyl)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CADTH ISSUES in EMERGING HEALTH TECHNOLOGIES Informing Decisions About New Health Technologies
CADTH ISSUES IN EMERGING HEALTH TECHNOLOGIES Informing Decisions About New Health Technologies Issue Mar 171 2018 Gene Therapy: An Overview of Approved and Pipeline Technologies CADTH ISSUES IN EMERGING HEALTH TECHNOLOGIES 1 Authors: Alison Sinclair, Saadul Islam, Sarah Jones Cite as: Gene therapy: an overview of approved and pipeline technologies. Ottawa: CADTH; 2018 Mar. (CADTH issues in emerging health technologies; issue 171). Acknowledgments: Louis de Léséleuc, Jeff Mason, Teo Quay, Joanne Kim, Lesley Dunfield, Eftyhia Helis, Iryna Magega, Jane Hurge ISSN: 1488-6324 (online) Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy- makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services. While CADTH has taken care to ensure that the information prepared by it in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. -

Spark Therapeutics and Pfizer Announce That SPK-9001, An
Spark Therapeutics and Pfizer Announce that SPK-9001, an Investigational Hemophilia B Medicine, has been Granted Access to the PRIority MEdicines (PRIME) Program by the European Medicines Agency PHILADELPHIA and NEW YORK, March 1, 2017 (GLOBE NEWSWIRE)— Spark Therapeutics (NASDAQ:ONCE) and Pfizer Inc. (NYSE:PFE) announced today that SPK-9001, the lead investigational candidate in the companies' SPK-FIX program has been granted support through the European Medicines Agency (EMA) PRIority MEdicines (PRIME) program. According to the EMA, PRIME is intended to enhance support for new treatments in development “that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.” “We are pleased that investigational SPK-9001 has been granted access to the PRIME program, which potentially will help accelerate the delivery of this potential gene therapy to European patients living with hemophilia B,” said Jeffrey D. Marrazzo, chief executive officer of Spark Therapeutics. “We appreciate the EMA’s dedication to moving treatments forward for patients who are underserved with current options, and we look forward to our continued collaboration on SPK-9001.” SPK-9001 is an experimental treatment for hemophilia B that is currently being investigated in an ongoing Phase 1/2 trial as a potential single-dose therapy. The PRIME program provides enhanced interaction and early dialogue with developers of promising medicines, which is intended to optimize development plans and speed up evaluation so these medicines can reach patients earlier. SPK-9001 received orphan product designation from the U.S. Food and Drug Administration (FDA) in September 2015, and later received breakthrough therapy designation from the FDA in July 2016. -

First Gene Therapy Fda-Approved for An
s FEATURE FIRST GENE THERAPY FDA-APPROVED FOR AN INHERITED RETINAL DISEASE The approval has stimulated research into gene therapies for other IRDs. BY MEGHAN J. DEBENEDICTIS, MS, LGC, MED, AND ALEKSANDRA V. RACHITSKAYA, MD he idea of gene therapy has been for clustered regularly interspaced short Scientists and companies have discussed in the medical litera- palindromic repeats) could allow edit- spent decades perfecting the use ture since as early as the 1970s. In ing of one or several sites within the of vectors for genetic material and 1972, Friedman and Roblin pro- mammalian genome.5 identifying ways to deliver them to posed that it was theoretically The most common type of gene increase therapeutic efficacy and possibleT to introduce “good” DNA to therapy is replacement gene therapy, treatment duration. Early investiga- replace defective DNA.1 Over the years, which involves replacing a mutated tions used viruses that delivered a number of gene therapy clinical trials gene that causes disease with a genes to every cell in the body, which emerged in efforts to treat genetic dis- healthy copy of that gene. It is first triggered a massive immune response eases of inborn errors of metabolism, necessary to identify the causative that could lead to organ failure. More all with varying degrees of success. mutated gene. This technology recently designed vectors deliver The basic principle of gene therapy works best in autosomal recessive specific genes to specific cells. is to put corrective genetic material biallelic disease with loss-of-function into cells to treat genetic disease. mutations. A vector is then created TARGET: IRDS Several gene therapy approaches, to carry a wild-type copy of the With the eye’s unique immunologic including replacement gene therapy, gene into the cell of interest. -

Landscape Review and Evidence Map of Gene Therapy, Part 2
EMERGING TECHNOLOGIES AND THERAPEUTICS REPORT 1 Landscape Review and Evidence Map of Gene Therapy, Part II: Chimeric Antigen Receptor- T cell (CAR-T), Autologous Cell, Antisense, RNA Interference (RNAi), Zinc Finger Nuclease (ZFN), Genetically Modified Oncolytic Herpes Virus Andrea Richardson, Eric Apaydin, Sangita Baxi, Jerry Vockley, Olamigoke Akinniranye, Rachel Ross, Jody Larkin, Aneesa Motala, Gulrez Azhar, and Susanne Hempel RAND Corporation June 2019 Revision [February 2020] Since the completion of this report a gene therapy herein described as AVXS-101 has been approved by the FDA as Zolgensma therefore the report was revised in November 2019 to reflect that change. All statements, findings, and conclusions in this publication are solely those of the authors and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI) or its Board of Governors. This publication was developed through a contract to support PCORI's work. Questions or comments may be sent to PCORI at [email protected] or by mail to Suite 900, 1828 L Street, NW, Washington, DC 20036. ©2019 Patient-Centered Outcomes Research Institute. For more information see https://www.pcori.org EMERGING TECHNOLOGIES AND THERAPEUTICS REPORT 2 Table of Contents Executive Summary ......................................................................................................... 6 Introduction of Report Series .......................................................................................... 8 History of Gene Therapies ........................................................................................... -

Luxturna: FDA Documents Reveal the Value of a Costly Gene Therapy
Drug Discovery Today Volume 24, Number 4 April 2019 PERSPECTIVE feature Luxturna: FDA documents reveal the value of a costly gene therapy Jonathan J. Darrow In 2017, the US Food and Drug Administration (FDA) approved voretigene neparvovec-rzyl (Luxturna), a gene therapy used to treat a rare form of inherited blindness. Widely described by the media as a curative treatment that ‘restores vision’, it was priced at US$850 000. Although voretigene neparvovec-rzyl represents a substantial therapeutic advance, most reports have failed to adequately describe study outcomes as documented by FDA reviewers. These documents reveal that the drug is not expected to restore normal vision, that only about half of treated patients met the FDA’s threshold for minimally meaningful improvement, that improvements might not persist long-term, that the most common PERSPECTIVE measure of visual function was rejected as a primary endpoint after yielding mixed results, and that two patients experienced permanent vision loss. Over US$100 million of additional publicly-funded costs are not evident from the US$850 000 figure. Features Introduction that targets a disease caused by mutations in a while looking through a tunnel. Other patients Voretigene neparvovec-rzyl (Luxturna) was ap- specific gene [1]. or their family members reported improvements proved by the FDA in December 2017 for the The transformative nature of voretigene such as the ability to read large print rather than treatment of biallelic retinal pigment epitheli- neparvovec-rzyl has been widely lauded by Braille, eat at dimly lit restaurants, use iPhone um-specific 65 kilodalton (RPE65) mutation-as- regulators, patients, and the media. -

Luxturna™ (Voretigene Neparvovec-Rzyl)
UnitedHealthcare® Commercial Medical Benefit Drug Policy Luxturna™ (Voretigene Neparvovec-Rzyl) Policy Number: 2020D0063F Effective Date: December 1, 2020 Instructions for Use Table of Contents Page Community Plan Policy Coverage Rationale ....................................................................... 1 • Luxturna™ (Voretigene Neparvovec-Rzyl) Documentation Requirements ...................................................... 1 Applicable Codes .......................................................................... 2 Background.................................................................................... 2 Benefit Considerations .................................................................. 2 Clinical Evidence ........................................................................... 3 U.S. Food and Drug Administration ............................................. 3 Centers for Medicare and Medicaid Services ............................. 3 References ..................................................................................... 3 Policy History/Revision Information ............................................. 4 Instructions for Use ....................................................................... 4 Coverage Rationale See Benefit Considerations Luxturna is proven and/or medically necessary for the treatment of Inherited Retinal Dystrophies (IRD) caused by mutations in the retinal pigment epithelium-specific protein 65kDa (RPE65) gene in patients who meet all of the following criteria:1-2 Patient is -

Spark Therapeutics Presents Updated Preliminary Data from Hemophilia
Spark Therapeutics Presents Updated Preliminary Data from Hemophilia B Phase 1/2 Trial Suggesting Consistent and Sustained Levels of Factor IX Activity at the Hemostasis and Thrombosis Research Society (HTRS) 2017 Scientific Symposium As of data cutoff, the annualized bleeding rate (ABR) has been reduced by 96 percent and the annualized infusion rate (AIR) reduced by 99 percent Both participants who began a tapering course of steroids have completed their regimen PHILADELPHIA, April 6, 2017 (GLOBE NEWSWIRE) — Spark Therapeutics (NASDAQ:ONCE) announced updated preliminary data today from 10 infused participants in the ongoing Phase 1/2 clinical trial of investigational SPK-9001 for hemophilia B. All participants have experienced consistent and sustained increases in factor IX activity following administration of the investigational therapy. These data will be presented at the Hemostasis and Thrombosis Research Society (HTRS) 2017 Scientific Symposium in Scottsdale, Arizona on Friday, April 7, by Adam Cuker, M.D., assistant professor of medicine at the Perelman School of Medicine of the University of Pennsylvania and a clinical investigator at Children’s Hospital of Philadelphia. Data as of March 24, 2017 will be presented on 10 participants in the study, who were dosed with a single administration of 5 x 1011 vector genomes (vg)/kg body weight. All participants have discontinued routine infusions of factor IX concentrates. Based on individual patient history prior to the study, ABR was reduced by 96 percent to a mean of 0.39 annual bleeds, compared with 9.2 bleeds before SPK-9001 administration. AIR was reduced 99 percent to a mean of 0.98 annual infusions, compared with 68.5 infusions before SPK-9001 administration. -

Fda Advisory Committee Briefing Document
Spark Therapeutics Briefing Document: October 12, 2017 FDA Advisory Committee Meeting FDA ADVISORY COMMITTEE BRIEFING DOCUMENT Spark Therapeutics, Inc LUXTURNATM (voretigene neparvovec) MEETING OF THE CELLULAR, TISSUE, AND GENE THERAPIES ADVISORY COMMITTEE MEETING DATE: October 12, 2017 ADVISORY COMMITTEE BRIEFING MATERIALS: AVAILABLE FOR PUBLIC RELEASE Page 1 of 149 Spark Therapeutics Briefing Document: October 12, 2017 FDA Advisory Committee Meeting TABLE OF CONTENTS Table of Contents ...........................................................................................................................2 List of Tables ..................................................................................................................................5 List of Figures .................................................................................................................................7 List of Abbreviations .....................................................................................................................9 1 Executive Summary ..................................................................................................11 1.1 Background and Unmet Need .....................................................................................11 1.2 Product Development Overview .................................................................................13 1.3 Natural History Study .................................................................................................14 1.4 Efficacy .......................................................................................................................16 -

Medical Policy #911 Gene Therapy for Inherited Retinal Dystrophy
Medical Policy Gene Therapy for Inherited Retinal Dystrophy Table of Contents • Policy: Commercial • Coding Information • Information Pertaining to All Policies • Policy: Medicare • Description • References • Authorization Information • Policy History • Endnotes Policy Number: 911 BCBSA Reference Number: 2.04.144 NCD/LCD: NA Related Policies None Policy Commercial Members: Managed Care (HMO and POS), PPO, and Indemnity Medicare HMO BlueSM and Medicare PPO BlueSM Members Preauthorization Request Form: Gene Therapy for Inherited Retinal Dystrophy This form must be completed and faxed to: Medical and Surgical: 1-888-282-0780; Medicare Advantage: 1-800-447-2994. Click here for Gene Therapy for Inherited Retinal Dystrophy Preauthorization Request Form, #926 Voretigene neparvovec-rzyl adeno-associated virus vector-based gene therapy subretinal injection is considered MEDICALLY NECESSARY for patients with vision loss due to biallelic RPE65 pathogenic or likely pathogenic1 variant-associated retinal dystrophy if they meet all of the following criteria: • Are adults (age <65 years) or children (age ≥3 years) • Documentation of the following: o Genetic testing confirming presence of bilallelic RPE65 pathogenic or likely pathogenic1 variant(s)* ▪ Single RPE65 pathogenic or likely pathogenic1 variant found in the homozygous state ▪ Two RPE65 pathogenic or likely pathogenic1 variants found in the trans configuration (compound heterozygous state) by segregation analysis o Presence of viable retinal cells as determined by treating physicians as assessed by optical coherence tomography imaging and/or ophthalmoscopy: ▪ An area of retina within the posterior pole of >100 μm thickness shown on optical coherence tomography, OR ▪ ≥3 disc areas of retina without atrophy or pigmentary degeneration within the posterior pole, OR 1 ▪ Any remaining visual field within 30° of fixation as measured by III4e/V4e isopter equivalent, OR ▪ Measureable full-field light sensitivity threshold (FST). -
Spark Therapeutics Presents Updated Interim Hemophilia B Data
Spark Therapeutics Presents Updated Interim Hemophilia B Data Supporting Consistent and Sustained Response at the International Society on Thrombosis and Haemostasis (ISTH) 2017 Congress Data demonstrate a 99-percent reduction in annualized infusion rate (AIR) and a 96-percent reduction in annualized bleeding rate (ABR) in 10 participants as of June 5, 2017 Five trial participants are now at least one-year post investigational SPK-9001 administration, including one participant out approximately 18 months; all have discontinued routine factor IX concentrate infusions and have sustained increases in factor IX activity levels PHILADELPHIA, July 10, 2017 -- Spark Therapeutics (NASDAQ: ONCE), a fully integrated gene therapy company dedicated to challenging the inevitability of genetic disease, announced today that 10 participants in its ongoing Phase 1/2 clinical trial of SPK-9001 for hemophilia B, as of the June 5, 2017 data cut off, had their AIR reduced approximately 99 percent to a mean of 1.0 annual infusion as of the data cut-off date, compared with 67.5 annual infusions before SPK-9001 administration. Nine of the 10 participants have not experienced a bleed since vector infusion; overall ABR was reduced by approximately 96 percent to a mean of 0.4 annual bleeds, compared with 11.1 bleeds before a single administration of 5 x 1011 vector genomes (vg)/kg body weight of SPK-9001. These data represent approximately 9.63 cumulative patient years of SPK-9001 exposure from the start of the trial, with one participant out approximately 18 months post-infusion and four additional participants out at least one year post-infusion. -

FDA Approves Spark Therapeutics' LUXTURNA
FDA Approves Spark Therapeutics’ LUXTURNA™ (voretigene neparvovec-rzyl), a One-time Gene Therapy for Patients with Confirmed Biallelic RPE65 Mutation-associated Retinal Dystrophy LUXTURNA is first gene therapy for a genetic disease, first and only pharmacologic treatment for an inherited retinal disease (IRD) and first adeno-associated virus (AAV) vector gene therapy approved in U.S. Children and adults living with IRD caused by biallelic RPE65 gene mutations nearly all progress to complete blindness Spark Therapeutics to offer comprehensive patient support services for eligible patients in U.S.; will share details on access and price in early January PHILADELPHIA, Dec. 19, 2017 (GLOBE NEWSWIRE) -- Spark Therapeutics (NASDAQ:ONCE), a fully integrated gene therapy company dedicated to challenging the inevitability of genetic disease, announced today that the U.S. Food and Drug Administration (FDA) has approved LUXTURNA™ (voretigene neparvovec- rzyl), a one-time gene therapy product indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. LUXTURNA should only be administered to patients with mutations on both copies of the RPE65 gene who have sufficient viable retinal cells as determined by their treating physicians. LUXTURNA is the first FDA-approved gene therapy for a genetic disease, the first and only pharmacologic treatment for an inherited retinal disease (IRD) and the first adeno-associated virus (AAV) vector gene therapy approved in the U.S. “Today’s landmark approval of LUXTURNA is a moment decades in the making for the field of gene therapy, the inherited retinal disease (IRD) community, and most importantly, patients with biallelic RPE65 mutation associated retinal dystrophy who now have the option to seek treatment,” said Jeffrey D. -

An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials
International Journal of Molecular Sciences Review An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials Wei Chiu 1,2,†, Ting-Yi Lin 2,3,† , Yun-Chia Chang 4 , Henkie Isahwan-Ahmad Mulyadi Lai 2,5 , Shen-Che Lin 1, Chun Ma 2,6, Aliaksandr A. Yarmishyn 2, Shiuan-Chen Lin 1,2, Kao-Jung Chang 1,2,7, Yu-Bai Chou 1,4, Chih-Chien Hsu 1,4 , Tai-Chi Lin 2,4, Shih-Jen Chen 2,4, Yueh Chien 2,8,* , Yi-Ping Yang 2,8,9,* and De-Kuang Hwang 2,4,* 1 School of Medicine, National Yang Ming Chiao Tung University, Taipei 11221, Taiwan; [email protected] (W.C.); [email protected] (S.-C.L.); [email protected] (S.-C.L.); [email protected] (K.-J.C.); [email protected] (Y.-B.C.); [email protected] (C.-C.H.) 2 Department of Medical Research, Taipei Veterans General Hospital, Taipei 11217, Taiwan; [email protected] (T.-Y.L.); [email protected] (H.I.-A.M.L.); [email protected] (C.M.); [email protected] (A.A.Y.); [email protected] (T.-C.L.); [email protected] (S.-J.C.) 3 School of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan 4 Department of Ophthalmology, Taipei Veterans General Hospital, Taipei 11217, Taiwan; [email protected] 5 Institute of Pharmacology, College of Medicine, National Yang Ming Chiao Tung University, Taipei 11221, Taiwan 6 Department of Medicine, National Taiwan University, Taipei 10617, Taiwan Citation: Chiu, W.; Lin, T.-Y.; Chang, 7 Institute of Clinical Medicine, National Yang Ming Chiao Tung University, Taipei 11221, Taiwan Y.-C.; Isahwan-Ahmad Mulyadi Lai, 8 Division of Basic Research, Department of Medical Research, Taipei Veterans General Hospital, H.; Lin, S.-C.; Ma, C.; Yarmishyn, Taipei 11217, Taiwan 9 A.A.; Lin, S.-C.; Chang, K.-J.; Chou, Institute of Food Safety and Health Risk Assessment, National Yang Ming Chiao Tung University, Taipei 11221, Taiwan Y.-B.; et al.