Rare Blood Group Variants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Other Blood Group Systems—Diego,Yt, Xg, Scianna, Dombrock
Review: other blood group systems—Diego, Yt, Xg, Scianna, Dombrock, Colton, Landsteiner- Wiener, and Indian K.M. B YRNE AND P.C. B YRNE Introduction Diego Blood Group System This review was prepared to provide a basic The Diego blood group system (ISBT: DI/010) has overview of “Other Blood Groups.” Some of the more expanded from its humble beginnings to now include major blood group systems, i.e., ABO, Rh, Kell, Duffy, up to 21 discrete antigens (Table 1). 3 Band 3, an anion and Kidd, are also reviewed in this issue and are not exchange, multi-pass membrane glycoprotein, is the covered here. The sheer mass of data on the MNS basic structure that carries the Diego system antigenic blood group system is so extensive and complicated determinants. 4 The gene that encodes the Band 3 that it justifies a review all of its own, and it is therefore protein is named SLC4A1 and its chromosomal location not discussed in this article. However, various aspects is 17q12–q21. 4 of MNS were described in recent papers in Di a and Di b are antithetical, resulting from a single Immunohematology. 1,2 nucleotide substitution (2561T>C) that gives rise to The blood group systems that are covered are those amino acid changes in the Band 3 protein (Leu854Pro). that most workers believe to have some degree of To date, the Di(a–b–) phenotype has not been clinical importance or interesting features: Diego (DI), described. The Di a and Di b antigens are resistant to Yt (YT), Xg (XG), Scianna (SC), Dombrock (DO), Colton treatment with the following enzymes/chemicals: (CO), Landsteiner-Wiener (LW), and Indian (IN). -

Genetic Screening for the Vel- Phenotype Circumvents Difficult Serological Screening Due to Variable Vel Expression Levels
UvA-DARE (Digital Academic Repository) Genetic basis of rare blood group variants Wigman, L. Publication date 2013 Link to publication Citation for published version (APA): Wigman, L. (2013). Genetic basis of rare blood group variants. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:28 Sep 2021 Chapter 6 Genetic screening for the Vel- phenotype circumvents difficult serological screening due to variable Vel expression levels Lonneke Haer-Wigman1 Shabnam Solati1 Aïcha Ait Soussan1 Erik Beckers2 Pim van der Harst3 Marga van Hulst-Sundermeijer4 Peter Ligthart1 Dick van Rhenen5 Hein Schepers3 Tamara Stegmann1 Masja de Haas1 C. Ellen van der Schoot1 1 Sanquin Research, Amsterdam and Landsteiner Laboratory, Academic Medical Centre, University of Amsterdam, Amsterdam, the Netherlands 2 Maastricht University Medical Centre+, Maastricht, The Netherlands 3 University Medical Center Groningen, University of Groningen, Groningen, The Netherlands 4 Sanquin Diagnostic Services, Amsterdam, The Netherlands 5 Sanquin Blood Bank, Rotterdam, Netherlands Manuscript under preparation Chapter 6 Abstract Background: Serological determination of the Vel- phenotype is challenging due to variable Vel expression levels. -

Genes and Human History
Genes and human history Gil McVean, Department of Statistics, Oxford Contact: [email protected] 2 3 • Where does the variation come from? • How old are the genetic differences between us? • Are these differences important? How different are our genomes? 5 Serological techniques for detecting variation Rabbit Anti-A antibodies Human A A B AB O 6 Blood group systems in humans • 28 known systems – 39 genes, 643 alleles System Genes Alleles Knops CR1 24+ ABO ABO 102 Landsteiner- ICAM4 3 Wiener Colton C4A, C4B 7+ Lewis FUT3, FUT6 14/20 Chido-rodgers AQP1 7 Lutheran LU 16 Colton DAF 10 MNS GYPA,GYPB,G 43 Diego SLC4A1 78 YPE Dombrock DO 9 OK BSG 2 Duffy FY 9 P-related A4GALT, 14/5 Gerbich GYPC 9 B3GALT3 GIL AQP3 2 RAPH-MER2 CD151 3 H/h FUT1, FUT2 27/22 Rh RHCE, RHD, 129 I GCNT2 7 RHAG Indian CD44 2 Scianna ERMAP 4 Kell KEL, XK 33/30 Xg XG, CD99 - Kidd SLC14A1 8 YT ACHE 4 http://www.bioc.aecom.yu.edu/bgmut/summary.htm 7 Protein electroporesis • Changes in mass/charge ratio resulting from amino acid substitutions in proteins can be detected Starch or agar gel -- +- + +- - - - -- - + - + +- -- Direction of travel • In humans, about 30% of all loci show polymorphism with a 6% chance of a pair of randomly drawn alleles at a locus being different Lewontin and Hubby (1966) Harris(1966) 8 The rise of DNA sequencing GATAAGACGGTGATACTCACGCGACGGGCTTGGGCGCCGACTCGTTCAGACGGTGACCCAACTTATCCGATCGACCC CGGGTCCCGATTTAGACTCGGTATCATTTCTGGTGATTATCGCCTGCAGGTTCAAGAACACGTTTGCAGCAAGAAGT GAGGGATTTTGTCAGTGATCCCAGTCTACGGAGCCAGTCACCTCTGGTAGTGAAATTTTATTCGTTCATCTTCATAT AAGTCGCAGACCGCACGATGGGGGACAGAATACTCGCACAGGAAGAACCGCGATGAACCGAGGTAACCTAACATCCT -

Blood Group Genomics
BLOOD GROUP GENOMICS Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of 36 blood group alleles among 396 Thai samples reveals region-specific variants Philaiphon Jongruamklang,1 Christoph Gassner,2 Stefan Meyer,2 Aksarakorn Kummasook,3 Marion Darlison,1 Chayanun Boonlum,4 Surin Chanta,5 Beat M. Frey,2 Martin L. Olsson,1,6* and Jill R. Storry 1,6* lood group antigen polymorphism shows great BACKGROUND: Blood group phenotype variation has variation in different world populations. The been attributed to potential resistance to pathogen reason for this is not completely understood; invasion. Variation was mapped in blood donors from however, it has been attributed to both Lampang (northern region) and Saraburi (central region), B Thailand, where malaria is endemic. The previously unknown blood group allele profiles were characterized ABBREVIATIONS: MALDI-TOF MS 5 matrix-assisted laser and the data were correlated with phenotypes. The high desorption/ionization time-of-flight mass spectrometry; PCR- incidence of the Vel-negative phenotype previously ASP 5 polymerase chain reaction with allele-specific reported in Thais was investigated. primers; SNP(s) 5 single nucleotide polymorphism(s). STUDY DESIGN AND METHODS: DNA from 396 From 1Hematology and Transfusion Medicine, Department of blood donors was analyzed by matrix-assisted laser Laboratory Medicine, Lund University, Lund, Sweden; desorption/ionization–time-of-flight mass spectrometry. 2Molecular Diagnostics & Research (MOC), Blood Transfusion Outliers were investigated by serology and DNA Service Zurich,€ Zurich-Schlieren,€ Switzerland; 3Department of sequencing. Allele discrimination assays for SMIM1 Medical Technology, School of Allied Health Sciences, rs1175550A/G and ACKR1 rs118062001C/T were University of Phayao, Phayao, Thailand; 4Transfusion Medicine, performed and correlated with antigen expression. -

Cemetery Inscriptions, Stark County, Ohio Are
!!l«^Siii«lii^lM«iil^if^ 0003055 ™ECHURCHoF JESUSCHRIST Permission to Microfilm ofL-MTER-DAY '^^'^ Famny History L.brary of Christ of C 'MN rrc Of The Church Jesus j/\llM I J Latter-aay Saints would iike permission lo preserve your material on microfilm anc make it avaiiabe to our Family History Centers If you agree, piease complete this cara and return it io us. authorize the Family History Library 'o micoiiim "he matenai named below and use this mic'ofilmed record as it seems most benefic a: n compi.ance with the Library s policies and proceoures I warrant that I am fuiiv authcze^ '3 O'cv ae :^ch permission ": e -I ma;e"a. ^^^^^W. 7" U)^ ro// STA/e,\ e^^vr/ c/V/?//-// OGS ll£& U/cr>7)i!t£.<rr yvf. 1- tv state ziD coae Si . ,J, PFGS293I 'p-aB =-'-3c-- -i^/ • CEMETERY INSCRIPTIONS Stark County, Ohio Volume VI CEMETERY INSCRIPTIONS STARK COUNTY. OHIO VOLUME VI INCLUDED IN VOLUME VI IS THE TOWNSHIP OF PERRY DATE MiCROFiCHED MAY I 8 1990 19l PrlOJCGT and G. S. FiGHS I* CALL # PREPARED BY THE MEMBERS OF THE STARK COUNTY CHAPTER THE OHIO GENEALOGICAL SOCIETY (^ OCTOBER 1. 1985 CHURCH , OF LATTER-DAY SA'.lM TS 11 FORWARD The contents of each volume of Cemetery Inscriptions, Stark County, Ohio are: Volume I: Townships of Lexington, Washington, Paris and Marlboro. Volume II: Townships of Nimishillen, Osnaburg, Sandy, Pike, Bethlehem and Sugar Creek. Volume III; Townships of Tuscarawas, Lawrence and Jackson. Volume IV: Lake Township and the cemeteries of Dead Man's Point and Forest Hill in Plain Township. -
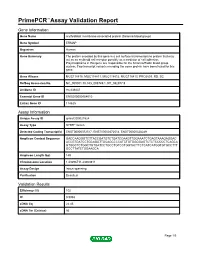
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name erythroblast membrane-associated protein (Scianna blood group) Gene Symbol ERMAP Organism Human Gene Summary The protein encoded by this gene is a cell surface transmembrane protein that may act as an erythroid cell receptor possibly as a mediator of cell adhesion. Polymorphisms in this gene are responsible for the Scianna/Radin blood group system. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases MGC118810, MGC118811, MGC118812, MGC118813, PRO2801, RD, SC RefSeq Accession No. NC_000001.10, NG_008749.1, NT_032977.9 UniGene ID Hs.439437 Ensembl Gene ID ENSG00000164010 Entrez Gene ID 114625 Assay Information Unique Assay ID qHsaCID0021524 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000372517, ENST00000372514, ENST00000328249 Amplicon Context Sequence GACCAAGGGTCTTACCGATGTCTGATCCAAGTTGGAAATCTGAGTAAAGAGGAC ACCGTGATCCTGCAGGTTGCAGCCCCATCTGTGGGGAGTCTCTCCCCCTCAGCA GTGGCTCTGGCTGTGATCCTGCCTGTCCTGGTACTTCTCATCATGGTGTGCCTTT GCCTTATCTGGAAGCA Amplicon Length (bp) 149 Chromosome Location 1:43296711-43300811 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 102 R2 0.9968 cDNA Cq 22.45 cDNA Tm (Celsius) 86 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 40.23 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report ERMAP, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference -

ERMAP Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Partial Recombinant ERMAP
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 ERMAP Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a partial recombinant ERMAP. Catalog # AT1943a Specification ERMAP Antibody (monoclonal) (M01) - Product Information Application WB, E Primary Accession Q96PL5 Other Accession NM_001017922 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 52605 ERMAP Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 114625 Immunogen (36.74 KDa) . Other Names Erythroid membrane-associated protein, hERMAP, Radin blood group antigen, Scianna blood group antigen, ERMAP, RD, SC Target/Specificity ERMAP (NP_001017922, 376 a.a. ~ 475 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Dilution WB~~1:500~1000 Format ERMAP monoclonal antibody (M01), clone Clear, colorless solution in phosphate 6F8 Western Blot analysis of ERMAP buffered saline, pH 7.2 . expression in HeLa ( (Cat # AT1943a ) Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Precautions ERMAP Antibody (monoclonal) (M01) is for research use only and not for use in diagnostic or therapeutic procedures. ERMAP Antibody (monoclonal) (M01) - Detection limit for recombinant GST tagged Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Protocols ERMAP is approximately 0.1ng/ml as a capture antibody. Provided below are standard protocols that you may find useful for product applications. ERMAP Antibody (monoclonal) (M01) - • Western Blot Background • Blocking Peptides • Dot Blot The protein encoded by this gene is a cell • Immunohistochemistry surface transmembrane protein that may act • Immunofluorescence as an erythroid cell receptor, possibly as a • Immunoprecipitation mediator of cell adhesion. -

Comprehensive Red Blood Cell and Platelet Antigen Prediction from Whole Genome Sequencing: Proof of Principle
BLOOD GROUP GENOMICS Comprehensive red blood cell and platelet antigen prediction from whole genome sequencing: proof of principle William J. Lane,1,2 Connie M. Westhoff,3 Jon Michael Uy,1 Maria Aguad,1 Robin Smeland-Wagman,1 Richard M. Kaufman,1 Heidi L. Rehm,1,2,4,5 Robert C. Green,2,5,6 and Leslie E. Silberstein7 for the MedSeq Project* rediction of red blood cell (RBC) and platelet BACKGROUND: There are 346 serologically defined (PLT) antigens using DNA assays has the poten- red blood cell (RBC) antigens and 33 serologically tial to augment or replace traditional serologic defined platelet (PLT) antigens, most of which have antigen typing in many situations. DNA-based known genetic changes in 45 RBC or six PLT genes that P typing methods are more easily automated, amenable to correlate with antigen expression. Polymorphic sites multiplexing, and do not require expensive and some- associated with antigen expression in the primary times difficult to obtain serologic immunoglobulin literature and reference databases are annotated according to nucleotide positions in cDNA. This makes antigen prediction from next-generation sequencing data ABBREVIATIONS: CDS 5 coding DNA sequence; NGS 5 challenging, since it uses genomic coordinates. next-generation sequencing; SNP(s) 5 single-nucleotide STUDY DESIGN AND METHODS: The conventional polymorphism(s); WGS 5 whole genome sequencing. cDNA reference sequences for all known RBC and PLT From the 1Department of Pathology, the 6Division of Genetics, genes that correlate with antigen expression were Department of Medicine, and the 7Division of Transfusion aligned to the human reference genome. The alignments Medicine, Department of Pathology, Brigham and Women’s allowed conversion of conventional cDNA nucleotide Hospital; and 2Harvard Medical School, Boston, Massachusetts; positions to the corresponding genomic coordinates. -

Clinical Significance of Antibodies to Antigens in the Raph, John Milton
R EVIEW Proceedings from the International Society of Blood Transfusion Working Party on Immunohaematology, Workshop on the Clinical Significance of Red Blood Cell Alloantibodies, September 2, 2016, Dubai Clinical significance of antibodies to antigens in the Raph, John Milton Hagen, I, Globoside, Gill, Rh-associated glycoprotein, FORS, JR, LAN, Vel, CD59, and Augustine blood group systems M. Moghaddam and A.A. Naghi This article reviews information on the clinical significance and 6 shared missense mutation c.511C>T (p.Argl71Cys) as of antibodies to antigens in the Raph, John Milton Hagen, I, well as a synonymous single-nucleotide mutation (c.579A>G) Globoside, Gill, Rh-associated glycoprotein, FORS, JR, LAN, Vel, and had no clinical features. Although the CD151 protein is CD59, and Augustine blood group systems. Antibodies to many of the antigens in these groups are rarely encountered because of the critical to cell adhesion and signaling and is implicated in high prevalence of the associated antigens in most populations. cancer progression, its significance in transfusion medicine is For many of these antibodies, the clinical significance—that is, limited to only one report of a hemolytic transfusion reaction the potential to cause reduced survival of transfused antigen- 3 positive red blood cells or a transfusion reaction (e.g., anti-P, (HTR). Least-incompatible RBC units should be selected anti-Jra, and anti-Lan), and/or hemolytic disease of the fetus and for transfusion to patients with anti-MER2.2 No information newborn (e.g., anti-RHAG4 and anti-Vel)—has been documented. on anti-MER2 causing hemolytic disease of the fetus and For other antibodies, their prevalence is so rare that information newborn (HDFN) is available.4 on the clinical significance of their antibodies is not available (e.g., anti-FORS1). -

Targeted Exome Sequencing Defines Novel and Rare Variants in Complex Blood Group Serology Cases for a Red Blood Cell Reference Laboratory Setting
IMMUNOHEMATOLOGY Targeted exome sequencing defines novel and rare variants in complex blood group serology cases for a red blood cell reference laboratory setting Elizna M. Schoeman ,1 Eileen V. Roulis ,1 Yew-Wah Liew,2 Jacqueline R. Martin,2 Tanya Powley,2 Brett Wilson,2 Glenda M. Millard ,1 Eunike C. McGowan ,1 Genghis H. Lopez ,1 Helen O’Brien ,1 Jennifer A. Condon,3 Robert L. Flower ,1 and Catherine A. Hyland 1 he key role of a modern red blood cell (RBC) ref- BACKGROUND: We previously demonstrated that erence laboratory in transfusion medicine is to targeted exome sequencing accurately defined blood employ serology to solve complex problems. group genotypes for reference panel samples Often, the lack of appropriate RBCs, sera, or characterized by serology and single-nucleotide T polymorphism (SNP) genotyping. Here we investigate reagents makes confident resolution of a complex case the application of this approach to resolve problematic difficult. Resources can be costly and scarce, if available at serology and SNP-typing cases. all, and often require collaboration and generosity of col- 1 STUDY DESIGN AND METHODS: The TruSight One leagues from other international laboratories. In addition, sequencing panel and MiSeq platform was used for reference laboratories commonly employ single- sequencing. CLC Genomics Workbench software was nucleotide polymorphism (SNP) typing microarrays for used for data analysis of the blood group genes RBC genotyping investigations; however, these arrays are implicated in the serology and SNP-typing problem. not comprehensive in their coverage of blood groups or 2-5 Sequence variants were compared to public databases variants. When the combination of traditional serologic listing blood group alleles. -

This Issue of Immunohematology Is Supported by a Contribution From
Journal of Blood Group Serology and Molecular Genetics VOLUME 33, N UMBER 4, 2017 This issue of Immunohematology is supported by a contribution from Grifols Diagnostics Solutions, Inc. Dedicated to advancement and education in molecular and serologic immunohematology Immunohematology Journal of Blood Group Serology and Molecular Genetics Volume 33, Number 4, 2017 CONTENTS O RIGINAL R EP O RT 147 Assessment of common red blood cell pretreatments to yield an accurate serologic antigen phenotype compared with genotype- predicted phenotype T. Horn, J. Hamilton, J. Kosanke, V.W. Hare, W. Kluver, W. Beres, S. Nance, and M.A. Keller C ASE R EP O RT 152 Anti-Vel alloimmunization and severe hemolytic disease of the fetus and newborn K.J. Moise Jr., Y. Morales, M.F. Bertholf, S.N. Rossmann, and Y. Bai S ER O LO GI C M ETH O D R EVIEW 155 Separation of multiple antibodies by adsorption with allogeneic red blood cells E.M. Ekema O RIGINAL R EP O RT 159 Hemovigilance and the Notify Library B.I. Whitaker, D.M. Strong, M.J. Gandhi, and E. Petrisli O RIGINAL R EP O RT 165 Clinical and laboratory profile of anti-M D. Basu, S. Basu, M. Reddy, K. Gupta, and M. Chandy S ER O LO GI C M ETH O D R EVIEW 170 Dithiothreitol treatment of red blood cells C.B. Bub C omm UNI C AT I O NS 173 Thank You S. Nance and C. Flickinger 174 175 183 187 191 E R R AT U M A NN O UN C E M ENTS A DVERTISE M ENTS I NSTRU C TI O NS S UBS C RIPTI O N F O R A UTH O RS I NF O R M AT I O N E DITO R - IN -C HIEF E DITO RIAL B OARD Sandra Nance, MS, MT(ASCP)SBB Philadelphia, Pennsylvania Patricia Arndt, MT(ASCP)SBB Geralyn M. -

Surname First Name Categorisation Abadin Jose Luis Silver Abbelen
2018 DRIVERS' CATEGORISATION LIST Updated on 09/07/2018 Drivers in red : revised categorisation Drivers in blue : new categorisation Surname First name Categorisation Abadin Jose Luis Silver Abbelen Klaus Bronze Abbott Hunter Silver Abbott James Silver Abe Kenji Bronze Abelli Julien Silver Abergel Gabriele Bronze Abkhazava Shota Bronze Abra Richard Silver Abreu Attila Gold Abril Vincent Gold Abt Christian Silver Abt Daniel Gold Accary Thomas Silver Acosta Hinojosa Julio Sebastian Silver Adam Jonathan Platinum Adams Rudi Bronze Adorf Dirk Silver Aeberhard Juerg Silver Afanasiev Sergei Silver Agostini Riccardo Gold Aguas Rui Gold Ahlin-Kottulinsky Mikaela Silver Ahrabian Darius Bronze Ajlani Karim Bronze Akata Emin Bronze Aksenov Stanislas Silver Al Faisal Abdulaziz Silver Al Harthy Ahmad Silver Al Masaood Humaid Bronze Al Qubaisi Khaled Bronze Al-Azhari Karim Bronze Alberico Neil Silver Albers Christijan Platinum Albert Michael Silver Albuquerque Filipe Platinum Alder Brian Silver Aleshin Mikhail Platinum Alesi Giuliano Silver Alessi Diego Silver Alexander Iradj Silver Alfaisal Saud Bronze Alguersuari Jaime Platinum Allegretta Vincent Silver Alleman Cyndie Silver Allemann Daniel Bronze Allen James Silver Allgàuer Egon Bronze Allison Austin Bronze Allmendinger AJ Gold Allos Manhal Bronze Almehairi Saeed Silver Almond Michael Silver Almudhaf Khaled Bronze Alon Robert Silver Alonso Fernando Platinum Altenburg Jeff Bronze Altevogt Peter Bronze Al-Thani Abdulrahman Silver Altoè Giacomo Silver Aluko Kolawole Bronze Alvarez Juan Cruz Silver Alzen