PAUP Tutorial.Pdf [437.0K]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 A) Login to the System B) Use the Appropriate Command to Determine Your Login Shell C) Use the /Etc/Passwd File to Verify the Result of Step B
CSE ([email protected] II-Sem) EXP-3 1 a) Login to the system b) Use the appropriate command to determine your login shell c) Use the /etc/passwd file to verify the result of step b. d) Use the ‘who’ command and redirect the result to a file called myfile1. Use the more command to see the contents of myfile1. e) Use the date and who commands in sequence (in one line) such that the output of date will display on the screen and the output of who will be redirected to a file called myfile2. Use the more command to check the contents of myfile2. 2 a) Write a “sed” command that deletes the first character in each line in a file. b) Write a “sed” command that deletes the character before the last character in each line in a file. c) Write a “sed” command that swaps the first and second words in each line in a file. a. Log into the system When we return on the system one screen will appear. In this we have to type 100.0.0.9 then we enter into editor. It asks our details such as Login : krishnasai password: Then we get log into the commands. bphanikrishna.wordpress.com FOSS-LAB Page 1 of 10 CSE ([email protected] II-Sem) EXP-3 b. use the appropriate command to determine your login shell Syntax: $ echo $SHELL Output: $ echo $SHELL /bin/bash Description:- What is "the shell"? Shell is a program that takes your commands from the keyboard and gives them to the operating system to perform. -
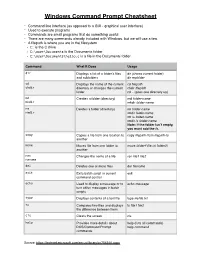
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

Don't Trust Traceroute (Completely)
Don’t Trust Traceroute (Completely) Pietro Marchetta, Valerio Persico, Ethan Katz-Bassett Antonio Pescapé University of Southern California, CA, USA University of Napoli Federico II, Italy [email protected] {pietro.marchetta,valerio.persico,pescape}@unina.it ABSTRACT In this work, we propose a methodology based on the alias resolu- tion process to demonstrate that the IP level view of the route pro- vided by traceroute may be a poor representation of the real router- level route followed by the traffic. More precisely, we show how the traceroute output can lead one to (i) inaccurately reconstruct the route by overestimating the load balancers along the paths toward the destination and (ii) erroneously infer routing changes. Categories and Subject Descriptors C.2.1 [Computer-communication networks]: Network Architec- ture and Design—Network topology (a) Traceroute reports two addresses at the 8-th hop. The common interpretation is that the 7-th hop is splitting the traffic along two Keywords different forwarding paths (case 1); another explanation is that the 8- th hop is an RFC compliant router using multiple interfaces to reply Internet topology; Traceroute; IP alias resolution; IP to Router to the source (case 2). mapping 1 1. INTRODUCTION 0.8 Operators and researchers rely on traceroute to measure routes and they assume that, if traceroute returns different IPs at a given 0.6 hop, it indicates different paths. However, this is not always the case. Although state-of-the-art implementations of traceroute al- 0.4 low to trace all the paths -

“Log” File in Stata
Updated July 2018 Creating a “Log” File in Stata This set of notes describes how to create a log file within the computer program Stata. It assumes that you have set Stata up on your computer (see the “Getting Started with Stata” handout), and that you have read in the set of data that you want to analyze (see the “Reading in Stata Format (.dta) Data Files” handout). A log file records all your Stata commands and output in a given session, with the exception of graphs. It is usually wise to retain a copy of the work that you’ve done on a given project, to refer to while you are writing up your findings, or later on when you are revising a paper. A log file is a separate file that has either a “.log” or “.smcl” extension. Saving the log as a .smcl file (“Stata Markup and Control Language file”) keeps the formatting from the Results window. It is recommended to save the log as a .log file. Although saving it as a .log file removes the formatting and saves the output in plain text format, it can be opened in most text editing programs. A .smcl file can only be opened in Stata. To create a log file: You may create a log file by typing log using ”filepath & filename” in the Stata Command box. On a PC: If one wanted to save a log file (.log) for a set of analyses on hard disk C:, in the folder “LOGS”, one would type log using "C:\LOGS\analysis_log.log" On a Mac: If one wanted to save a log file (.log) for a set of analyses in user1’s folder on the hard drive, in the folder “logs”, one would type log using "/Users/user1/logs/analysis_log.log" If you would like to replace an existing log file with a newer version add “replace” after the file name (Note: PC file path) log using "C:\LOGS\analysis_log.log", replace Alternately, you can use the menu: click on File, then on Log, then on Begin. -

Basic STATA Commands
Summer School on Capability and Multidimensional Poverty OPHI-HDCA, 2011 Oxford Poverty and Human Development Initiative (OPHI) http://ophi.qeh.ox.ac.uk Oxford Dept of International Development, Queen Elizabeth House, University of Oxford Basic STATA commands Typical STATA window Review Results Variables Commands Exploring your data Create a do file doedit Change your directory cd “c:\your directory” Open your database use database, clear Import from Excel (csv file) insheet using "filename.csv" Condition (after the following commands) if var1==3 or if var1==”male” Equivalence symbols: == equal; ~= not equal; != not equal; > greater than; >= greater than or equal; < less than; <= less than or equal; & and; | or. Weight [iw=weight] or [aw=weight] Browse your database browse Look for a variables lookfor “any word/topic” Summarize a variable (mean, standard su variable1 variable2 variable3 deviation, min. and max.) Tabulate a variable (per category) tab variable1 (add a second variables for cross tabs) Statistics for variables by subgroups tabstat variable1 variable2, s(n mean) by(group) Information of a variable (coding) codebook variable1, tab(99) Keep certain variables (use drop for the keep var1 var2 var3 opposite) Save a dataset save filename, [replace] Summer School on Capability and Multidimensional Poverty OPHI-HDCA, 2011 Creating Variables Generate a new variable (a number or a gen new_variable = 1 combinations of other variables) gen new_variable = variable1+ variable2 Generate a new variable conditional gen new_variable -

Forest Quickstart Guide for Linguists
Forest Quickstart Guide for Linguists Guido Vanden Wyngaerd [email protected] June 28, 2020 Contents 1 Introduction 1 2 Loading Forest 2 3 Basic Usage 2 4 Adjusting node spacing 4 5 Triangles 7 6 Unlabelled nodes 9 7 Horizontal alignment of terminals 10 8 Arrows 11 9 Highlighting 14 1 Introduction Forest is a package for drawing linguistic (and other) tree diagrams de- veloped by Sašo Živanović. This manual provides a quickstart guide for linguists with just the essential things that you need to get started. More 1 extensive documentation is available from the CTAN-archive. Forest is based on the TikZ package; more information about its commands, in par- ticular those controlling the appearance of the nodes, the arrows, and the highlighting can be found in the TikZ documentation. 2 Loading Forest In your preamble, put \usepackage[linguistics]{forest} The linguistics option makes for nice trees, in which the branches meet above the two nodes that they join; it will also align the example number (provided by linguex) with the top of the tree: (1) CP C IP I VP V NP 3 Basic Usage Forest uses a familiar labelled brackets syntax. The code below will out- put the tree in (1) above (\ex. requires the linguex package and provides the example number): \ex. \begin{forest} [CP[C][IP[I][VP[V][NP]]]] \end{forest} Forest will parse the above code without problem, but you are likely to soon get lost in your labelled brackets with more complicated trees if you write the code this way. The better alternative is to arrange the nodes over multiple lines: 2 \ex. -

NETSTAT Command
NETSTAT Command | NETSTAT Command | Use the NETSTAT command to display network status of the local host. | | ┌┐────────────── | 55──NETSTAT─────6─┤ Option ├─┴──┬────────────────────────────────── ┬ ─ ─ ─ ────────────────────────────────────────5% | │┌┐───────────────────── │ | └─(──SELect───6─┤ Select_String ├─┴ ─ ┘ | Option: | ┌┐─COnn────── (1, 2) ──────────────── | ├──┼─────────────────────────── ┼ ─ ──────────────────────────────────────────────────────────────────────────────┤ | ├─ALL───(2)──────────────────── ┤ | ├─ALLConn─────(1, 2) ────────────── ┤ | ├─ARp ipaddress───────────── ┤ | ├─CLients─────────────────── ┤ | ├─DEvlinks────────────────── ┤ | ├─Gate───(3)─────────────────── ┤ | ├─┬─Help─ ┬─ ───────────────── ┤ | │└┘─?──── │ | ├─HOme────────────────────── ┤ | │┌┐─2ð────── │ | ├─Interval─────(1, 2) ─┼───────── ┼─ ┤ | │└┘─seconds─ │ | ├─LEVel───────────────────── ┤ | ├─POOLsize────────────────── ┤ | ├─SOCKets─────────────────── ┤ | ├─TCp serverid───(1) ─────────── ┤ | ├─TELnet───(4)───────────────── ┤ | ├─Up──────────────────────── ┤ | └┘─┤ Command ├───(5)──────────── | Command: | ├──┬─CP cp_command───(6) ─ ┬ ────────────────────────────────────────────────────────────────────────────────────────┤ | ├─DELarp ipaddress─ ┤ | ├─DRop conn_num──── ┤ | └─RESETPool──────── ┘ | Select_String: | ├─ ─┬─ipaddress────(3) ┬ ─ ───────────────────────────────────────────────────────────────────────────────────────────┤ | ├─ldev_num─────(4) ┤ | └─userid────(2) ─── ┘ | Notes: | 1 Only ALLCON, CONN and TCP are valid with INTERVAL. | 2 The userid -

PC23 and PC43 Desktop Printer User Manual Document Change Record This Page Records Changes to This Document
PC23 | PC43 Desktop Printer PC23d, PC43d, PC43t User Manual Intermec by Honeywell 6001 36th Ave.W. Everett, WA 98203 U.S.A. www.intermec.com The information contained herein is provided solely for the purpose of allowing customers to operate and service Intermec-manufactured equipment and is not to be released, reproduced, or used for any other purpose without written permission of Intermec by Honeywell. Information and specifications contained in this document are subject to change without prior notice and do not represent a commitment on the part of Intermec by Honeywell. © 2012–2014 Intermec by Honeywell. All rights reserved. The word Intermec, the Intermec logo, Fingerprint, Ready-to-Work, and SmartSystems are either trademarks or registered trademarks of Intermec by Honeywell. For patent information, please refer to www.hsmpats.com Wi-Fi is a registered certification mark of the Wi-Fi Alliance. Microsoft, Windows, and the Windows logo are registered trademarks of Microsoft Corporation in the United States and/or other countries. Bluetooth is a trademark of Bluetooth SIG, Inc., U.S.A. The products described herein comply with the requirements of the ENERGY STAR. As an ENERGY STAR partner, Intermec Technologies has determined that this product meets the ENERGY STAR guidelines for energy efficiency. For more information on the ENERGY STAR program, see www.energystar.gov. The ENERGY STAR does not represent EPA endorsement of any product or service. ii PC23 and PC43 Desktop Printer User Manual Document Change Record This page records changes to this document. The document was originally released as Revision 001. Version Number Date Description of Change 005 12/2014 Revised to support MR7 firmware release. -

Introduction to Unix Shell
Introduction to Unix Shell François Serra, David Castillo, Marc A. Marti- Renom Genome Biology Group (CNAG) Structural Genomics Group (CRG) Run Store Programs Data Communicate Interact with each other with us The Unix Shell Introduction Interact with us Rewiring Telepathy Typewriter Speech WIMP The Unix Shell Introduction user logs in The Unix Shell Introduction user logs in user types command The Unix Shell Introduction user logs in user types command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off shell The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output -

11 Creating New Variables Generate and Replace This Chapter Shows the Basics of Creating and Modifying Variables in Stata
11 Creating new variables generate and replace This chapter shows the basics of creating and modifying variables in Stata. We saw how to work with the Data Editor in [GSM] 6 Using the Data Editor—this chapter shows how we would do this from the Command window. The two primary commands used for this are • generate for creating new variables. It has a minimum abbreviation of g. • replace for replacing the values of an existing variable. It may not be abbreviated because it alters existing data and hence can be considered dangerous. The most basic form for creating new variables is generate newvar = exp, where exp is any kind of expression. Of course, both generate and replace can be used with if and in qualifiers. An expression is a formula made up of constants, existing variables, operators, and functions. Some examples of expressions (using variables from the auto dataset) would be 2 + price, weight^2 or sqrt(gear ratio). The operators defined in Stata are given in the table below: Relational Arithmetic Logical (numeric and string) + addition ! not > greater than - subtraction | or < less than * multiplication & and >= > or equal / division <= < or equal ^ power == equal != not equal + string concatenation Stata has many mathematical, statistical, string, date, time-series, and programming functions. See help functions for the basics, and see[ D] functions for a complete list and full details of all the built-in functions. You can use menus and dialogs to create new variables and modify existing variables by selecting menu items from the Data > Create or change data menu. This feature can be handy for finding functions quickly. -

Generating Define.Xml Using SAS® by Element-By-Element and Domain-By-Domian Mechanism Lina Qin, Beijing, China
PharmaSUG China 2015 - 30 Generating Define.xml Using SAS® By Element-by-Element And Domain-by-Domian Mechanism Lina Qin, Beijing, China ABSTRACT An element-by-element and domain-by-domain mechanism is introduced for generating define.xml using SAS®. Based on CDISC Define-XML Specification, each element in define.xml can be generated by applying a set of templates instead of writing a great deal of “put” statements. This will make programs more succinct and flexible. The define.xml file can be generated simply by combining, in proper order, all of the relevant elements. Moreover, as each element related to a certain domain can be separately created, it is possible to generate a single-domain define.xml by combining all relevant elements of that domain. This mechanism greatly facilitates generating and validating define.xml. Keywords: Define-XML, define.xml, CRT-DDS, SDTM, CDISC, SAS 1 INTRODUCTION Define.xml file is used to describe CDISC SDTM data sets for the purpose of submissions to FDA[1]. While the structure of define.xml is set by CDISC Define-XML Specification[1], its content is subject to specific clinical study. Many experienced SAS experts have explored different methods to generate define.xml using SAS[5][6][7][8][9]. This paper will introduce a new method to generate define.xml using SAS. The method has two features. First, each element in define.xml can be generated by applying a set of templates instead of writing a great deal of “put” statements. This will make programs more succinct and flexible. Second, the define.xml can be generated on a domain-by-domain basis, which means each domain can have its own separate define.xml file. -

Linux Terminal Commands Man Pwd Ls Cd Olmo S
Python Olmo S. Zavala Romero Welcome Computers File system Super basics of Linux terminal Commands man pwd ls cd Olmo S. Zavala Romero mkdir touch Center of Atmospheric Sciences, UNAM rm mv Ex1 August 9, 2017 Regular expres- sions grep Ex2 More Python Olmo S. 1 Welcome Zavala Romero 2 Computers Welcome 3 File system Computers File system 4 Commands Commands man man pwd pwd ls ls cd mkdir cd touch mkdir rm touch mv Ex1 rm Regular mv expres- sions Ex1 grep Regular expressions Ex2 More grep Ex2 More Welcome to this course! Python Olmo S. Zavala Romero Welcome Computers File system Commands 1 Who am I? man pwd 2 Syllabus ls cd 3 Intro survey https://goo.gl/forms/SD6BM6KHKRlDOpZx1 mkdir 4 touch Homework 1 due this Sunday. rm mv Ex1 Regular expres- sions grep Ex2 More How does a computer works? Python Olmo S. Zavala Romero Welcome Computers File system Commands man pwd ls cd mkdir touch rm mv Ex1 Regular 1 CPU Central Processing Unit. All the computations happen there. expres- sions 2 HD Hard Drive. Stores persistent information in binary format. grep 3 RAM Random Access Memory. Faster memory, closer to the CPU, not persistent. Ex2 More 4 GPU Graphics Processing Unit. Used to process graphics. https://teentechdaily.files.wordpress.com/2015/06/computer-parts-diagram.jpg What is a file? Python Olmo S. Zavala Romero Welcome Computers File system Commands man pwd ls cd mkdir touch rm mv Section inside the HD with 0’s and 1’s. Ex1 Regular File extension is used to identify the meaning of those 0’s and 1’s.