One Thousand Plant Transcriptomes and the Phylogenomics of Green Plants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FLAME ACANTHUS OR HUMMINGBIRD BUSH Anisacanthus Quadrifidus Var
FLAME ACANTHUS OR HUMMINGBIRD BUSH Anisacanthus quadrifidus var. wrightii (A. wrightii) Characteristics Type: Perennial Maintenance: Low Zone: 7 to 10 Flower: Showy Height: 3.00 to 5.00 feet Attracts: Hummingbirds, Butterflies Spread: 3.00 to 4.00 feet Other: Winter Interest Bloom Time: June to September Tolerate: Deer, Drought, Clay Soil, Bloom Description: Reddish orange Shallow-Rocky Soil Sun: Full sun Texas Native Water: Dry Culture From midsummer through frost, flame acanthus is covered with long, slender, red or orange blooms that hummingbirds love. It is a drought tolerant, heat-loving small shrub that works as well in the perennial border as it does as an informal hedge or specimen plant. The bark is light and flaky and makes an interesting winter and early spring accent. Flame acanthus is late to come out in the spring, and benefits from periodic shearing or even severe cutting back in early spring. It grows in the Edwards Plateau on rocky banks and floodplains, but is adaptable to sunny, well-drained exposures throughout the state, even Houston. It is a good choice for sites with poor soils and reflected heat - although supplemental water in dry summer months will encourage flowering. Best grown in medium to dry, well-draining soils in full sun, but is adaptable to many soil types including poor, rocky soils and heavy, clay soils. Tolerant of drought, and takes well to pot culture. Noteworthy Characteristics Anisacanthus quadrifidus var. wrightii, is an upright, deciduous shrub reaching up to 5' tall and 4' wide with an informal, spreading appearance. It is native to extreme south-central Texas and adjacent northern Mexico, where it is found growing on rocky, calcareous slopes and floodplains. -

Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi
The University of Southern Mississippi The Aquila Digital Community Honors Theses Honors College Spring 5-2016 Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi Hanna M. Miller University of Southern Mississippi Follow this and additional works at: https://aquila.usm.edu/honors_theses Part of the Biodiversity Commons, and the Botany Commons Recommended Citation Miller, Hanna M., "Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi" (2016). Honors Theses. 389. https://aquila.usm.edu/honors_theses/389 This Honors College Thesis is brought to you for free and open access by the Honors College at The Aquila Digital Community. It has been accepted for inclusion in Honors Theses by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. The University of Southern Mississippi Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi by Hanna Miller A Thesis Submitted to the Honors College of The University of Southern Mississippi in Partial Fulfillment of the Requirement for the Degree of Bachelor of Science in the Department of Biological Sciences May 2016 ii Approved by _________________________________ Mac H. Alford, Ph.D., Thesis Adviser Professor of Biological Sciences _________________________________ Shiao Y. Wang, Ph.D., Chair Department of Biological Sciences _________________________________ Ellen Weinauer, Ph.D., Dean Honors College iii Abstract The North American Coastal Plain contains some of the highest plant diversity in the temperate world. However, most of the region has remained unstudied, resulting in a lack of knowledge about the unique plant communities present there. -

A New Application for Phylogenetic Marker Development Using Angiosperm Transcriptomes Author(S): Srikar Chamala, Nicolás García, Grant T
MarkerMiner 1.0: A New Application for Phylogenetic Marker Development Using Angiosperm Transcriptomes Author(s): Srikar Chamala, Nicolás García, Grant T. Godden, Vivek Krishnakumar, Ingrid E. Jordon- Thaden, Riet De Smet, W. Brad Barbazuk, Douglas E. Soltis, and Pamela S. Soltis Source: Applications in Plant Sciences, 3(4) Published By: Botanical Society of America DOI: http://dx.doi.org/10.3732/apps.1400115 URL: http://www.bioone.org/doi/full/10.3732/apps.1400115 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. ApApplicatitionsons Applications in Plant Sciences 2015 3 ( 4 ): 1400115 inin PlPlant ScienSciencesces S OFTWARE NOTE M ARKERMINER 1.0: A NEW APPLICATION FOR PHYLOGENETIC 1 MARKER DEVELOPMENT USING ANGIOSPERM TRANSCRIPTOMES S RIKAR C HAMALA 2,12 , N ICOLÁS G ARCÍA 2,3,4 * , GRANT T . G ODDEN 2,3,5 * , V IVEK K RISHNAKUMAR 6 , I NGRID E. -

Ardisia Humilis Vahl.) Planting Condition Toward the Alpha-Glucosidase Inhibition Activity in Vitro
Pharmacogn J. 2020; 12(2): 377-385 A Multifaceted Journal in the field of Natural Products and Pharmacognosy Research Article www.phcogj.com Study of the Effect of Lampeni (Ardisia humilis Vahl.) Planting Condition toward the Alpha-glucosidase Inhibition Activity In vitro Sri Ningsih1,*, Fifit Juniarti1, Idah Rosidah1, Adam Arditya Fajriawan1, Kurnia Agustini1, Syofi Rosmalawati2, Agung Eru Wibowo2, Erliana Sasikirana3, Wahono Sumaryono3 ABSTRACT Background: The quality of a medicinal plant is influenced by agronomic conditions and harvesting time. Objective: This study aimed to evaluate the effect of planting method (open- air (OA) and shedding house (SH)) and harvesting time (2, 4, 6 months) of Lampeni (Ardisia Sri Ningsih1,*, Fifit Juniarti1, humilis Vahl.) toward the inhibitory activity of alpha-glucosidase. Methods: The Lampeni Idah Rosidah1, Adam Arditya seedling were placed under controlled light conditions (SH) and on direct sun exposure (OA). Fajriawan1, Kurnia Agustini1, Harvesting of the leaves was carried out at the age of 2, 4, and 6 months after plantation Syofi Rosmalawati2, Agung Eru (2m, 4m, and 6m). Each leaves dry powder was refluxed with methanol 70% and followed Wibowo2, Erliana Sasikirana3, by liquid-liquid partition using n-hexane, ethyl acetate (EtOAc), and water. All samples were Wahono Sumaryono3 evaluated toward inhibition of the alpha-glucosidase enzyme in vitro. Total phenol levels were determined using Folin-Ciocalteu reagent. Results: The results showed that EtOAc fractions 1Center for Pharmaceutical and Medical Technology, Agency for the Assessment and of both plantation techniques exhibited the highest inhibition of alpha-glucosidase. The highest Application of Technology. Laptiab building, activity was demonstrated by the 4m-OA-EtOAc fraction (IC50, 93.50 ppm) and followed by Puspiptek Serpong Area, South Tangerang, the 6m-OA-EtOAc fraction (IC50, 98.13 ppm). -

The Ecology and Biogeography of Athrotaxis D. Don
THE ECOLOGY AND BIOGEOGRAPHY OF ATHROTAXIS D. DON. ,smo,e5 by Philip J. Cullen B.Sc. (Forestry) submitted in fulfilment of the requirements for the degree of Master of Science UNIVERSITY OF TASMANIA HOBART February 1987 DECLARATION This thesis contains no material which has been submitted for the award of any other degree or diploma in any university and contains no copy or paraphrases of material previously published or writtern by another person, except where due reference is made in the text. P. J. Cullen. CONTENTS PAGE ACKNOWLEGEMENTS ABSTRACT II LIST OF FIGURES IV LIST OF TABLES VI LIST OF PLATES VIII CHAPTER 1: INTRODUCTION , 1 CHAPTER 2: DISTRIBUTION AND SYNECOLOGY 2.1 Introduction 8 2.2 Methods 14 2.3 Results and Discussion 2.3.1 Stand Classification 19 2.3.2 Stand Ordination 32 CHAPTER 3: THE REGENERATION MODES OF ATHROTAXIS CUPRESSOIDES AND ATHROTAXIS SELAGINOIDES 3.1 Introduction 38 3.2 Methods 3.2.1 Stand demography 40 3.2.2 Spatial distribution of seedlings 43 3.2.3 Seed dispersal 44 3.2.4 Vegetative reproduction 45 3.3 Results and Discussion 3.3.1 Size/age correlations 48 3.3.2 Seed production and dispersal 52 3.3.3 Vegetative regeneration 55 3.3.4 Stand demography and seedling distribution of Athrotaxis cupressoides 60 3.3.5 Stand demography and seedling distribution of Athrotaxis seiaginoides 82 3.3.6 The presence of Athrotaxis laxifolia . 88 3.4 Conclusion 89 CHAPTER THE RELATIVE FROST RESISTANCE OF ATHROTAXIS CUPRESSOIDES AND ATHROTAXIS SELAGINOIDES SEEDLINGS 4.1 Introduction 92 4.2 Methods 93 4,3 Results 96 4.4 Discussion 99 CHAPTER 5: THE EFFECT OF GRAZING ON THE SEEDLING REGENERATION OF ATHROTAXIS CUPRESSOIDES 5.1 Introduction 107 5.2 Methods 107 5.3 Results 109 5.4 Discussion 111 CHAPTER 6: DISCUSSION 116 REFERENCES 135 APPENDIX A: REGENERATION PATTERNS IN POPULATIONS OF ATHROTAXIS SELAGINOIDES D. -

Phenotypic Landscape Inference Reveals Multiple Evolutionary Paths to C4 Photosynthesis
RESEARCH ARTICLE elife.elifesciences.org Phenotypic landscape inference reveals multiple evolutionary paths to C4 photosynthesis Ben P Williams1†, Iain G Johnston2†, Sarah Covshoff1, Julian M Hibberd1* 1Department of Plant Sciences, University of Cambridge, Cambridge, United Kingdom; 2Department of Mathematics, Imperial College London, London, United Kingdom Abstract C4 photosynthesis has independently evolved from the ancestral C3 pathway in at least 60 plant lineages, but, as with other complex traits, how it evolved is unclear. Here we show that the polyphyletic appearance of C4 photosynthesis is associated with diverse and flexible evolutionary paths that group into four major trajectories. We conducted a meta-analysis of 18 lineages containing species that use C3, C4, or intermediate C3–C4 forms of photosynthesis to parameterise a 16-dimensional phenotypic landscape. We then developed and experimentally verified a novel Bayesian approach based on a hidden Markov model that predicts how the C4 phenotype evolved. The alternative evolutionary histories underlying the appearance of C4 photosynthesis were determined by ancestral lineage and initial phenotypic alterations unrelated to photosynthesis. We conclude that the order of C4 trait acquisition is flexible and driven by non-photosynthetic drivers. This flexibility will have facilitated the convergent evolution of this complex trait. DOI: 10.7554/eLife.00961.001 Introduction *For correspondence: Julian. The convergent evolution of complex traits is surprisingly common, with examples including camera- [email protected] like eyes of cephalopods, vertebrates, and cnidaria (Kozmik et al., 2008), mimicry in invertebrates and †These authors contributed vertebrates (Santos et al., 2003; Wilson et al., 2012) and the different photosynthetic machineries of equally to this work plants (Sage et al., 2011a). -

Pdf 755.65 K
Trends Phytochem. Res. 4(4) 2020 177-192 Trends in Phytochemical Research (TPR) Journal Homepage: http://tpr.iau-shahrood.ac.ir Original Research Article Chemical composition, insect antifeeding, insecticidal, herbicidal, antioxidant and anti-inflammatory potential of Ardisia solonaceae Roxb. root extract BAHAAR ANJUM1, RAVENDRA KUMAR1 *, RANDEEP KUMAR1, OM PRAKASH1, R.M. SRIVASTAVA2, D.S. RAWAT3 AND A.K. PANT1 1Department of Chemistry, College of Basic Sciences and Humanities, G.B. Pant University of Agriculture and Technology, Pantnagar-263145, U.S. Nagar, Uttarakhand, India 2Department of Entomology, College of Agriculture, G.B. Pant University of Agriculture and Technology, Pantnagar-263145, U.S. Nagar, Uttarakhand, India 3Department of Biological Sciences, College of Basic Sciences and Humanities, G.B. Pant University of Agriculture and Technology, Pantnagar-263145, U.S. Nagar, Uttarakhand, India ABSTRACT ARTICLE HISTORY The objectives of this research were to investigate the qualitative and quantitative analysis Received: 19 March 2020 of Ethyl Acetate Root Extract of Ardisia solanacea Roxb. (EREAS) and estimation of its Revised: 08 June 2020 biological activities. Phytochemical screening of EREAS showed the abundance of total Accepted: 05 October 2020 phenolics, flavanoids, ortho-dihydric phenols, alkaloids, diterpenes and triterpenes etc. The ePublished: 02 December 2020 quantitative analysis of EREAS was also carried out by GC/MS and α-amyrenone (13.3%) was found to be the major component. Antifeeding activity monitored through no choice leaf dip method against Spilosoma oblique. The results revealed dose and time dependent KEYWORDS antifeeding activity, where the 100% mortality was observed signifying the intense insecticidal activity. The herbicidal activity of extracts was evaluated against the Raphanus raphanistrum α-Amyrenone seeds. -

1995 New Zealand Botanical Society
NEW ZEALAND BOTANICAL SOCIETY NEWSLETTER NUMBER 42 DECEMBER 1995 New Zealand Botanical Society President: Jessica Beever Secretary/Treasurer: Anthony Wright Committee: Catherine Beard, Colin Webb, Carol West, Beverley Clarkson, Bruce Clarkson Address: C/- Auckland Institute & Museum Private Bag 92018 AUCKLAND Subscriptions The 1996 ordinary and institutional subs are $14 (reduced to $10 if paid by the due date on the subscription invoice). The 1996 student sub, available to full-time students, is $7 (reduced to $5 if paid by the due date on the subscription invoice). Back issues of the Newsletter are available at $2.50 each - from Number 1 (August 1985) to Number 41 (September 1995). Since 1986 the Newsletter has appeared quarterly in March, June, September and December. New subscriptions are always welcome and these, together with back issue orders, should be sent to the Secretary/Treasurer (address above). Subscriptions are due by 28 February of each year for that calendar year. Existing subscribers are sent an invoice with the December Newsletter for the next year's subscription which offers a reduction if this is paid by the due date. If you are in arrears with your subscription a reminder notice comes attached to each issue of the Newsletter. Deadline for next issue The deadline for the March 1996 issue (Number 43) is 28 February 1996. Please forward contributions to: Bruce & Beverley Clarkson, Editors NZ Botanical Society Newsletter 7 Lynwood Place HAMILTON Contributions may be provided on floppy disc (preferably in Word Perfect 5.1) or by e-mail ([email protected]). NEW ZEALAND BOTANICAL SOCIETY NEWSLETTER NUMBER 42 DECEMBER 1995 CONTENTS News New Zealand Botanical Society News From the Secretary 2 Regional Botanical Society News Nelson Botanical Society . -

Astereae, Asteraceae) Using Molecular Phylogeny of ITS
Turkish Journal of Botany Turk J Bot (2015) 39: 808-824 http://journals.tubitak.gov.tr/botany/ © TÜBİTAK Research Article doi:10.3906/bot-1410-12 Relationships and generic delimitation of Eurasian genera of the subtribe Asterinae (Astereae, Asteraceae) using molecular phylogeny of ITS 1, 2,3 4 Elena KOROLYUK *, Alexey MAKUNIN , Tatiana MATVEEVA 1 Central Siberian Botanical Garden, Siberian Branch of Russian Academy of Sciences, Novosibirsk, Russia 2 Institute of Molecular and Cell Biology, Siberian Branch of Russian Academy of Sciences, Novosibirsk, Russia 3 Theodosius Dobzhansky Center for Genome Bioinformatics, Saint Petersburg State University, Saint Petersburg, Russia 4 Department of Genetics & Biotechnology, Saint Petersburg State University, Saint Petersburg, Russia Received: 12.10.2014 Accepted/Published Online: 02.04.2015 Printed: 30.09.2015 Abstract: The subtribe Asterinae (Astereae, Asteraceae) includes highly variable, often polyploid species. Recent findings based on molecular methods led to revision of its volume. However, most of these studies lacked species from Eurasia, where a lot of previous taxonomic treatments of the subtribe exist. In this study we used molecular phylogenetics methods with internal transcribed spacer (ITS) as a marker to resolve evolutionary relations between representatives of the subtribe Asterinae from Siberia, Kazakhstan, and the European part of Russia. Our reconstruction revealed that a clade including all Asterinae species is paraphyletic. Inside this clade, there are species with unresolved basal positions, for example Erigeron flaccidus and its relatives. Moreover, several well-supported groups exist: group of the genera Galatella, Crinitaria, Linosyris, and Tripolium; group of species of North American origin; and three related groups of Eurasian species: typical Eurasian asters, Heteropappus group (genera Heteropappus, Kalimeris), and Asterothamnus group (genera Asterothamnus, Rhinactinidia). -

A Legacy of Plants N His Short Life, Douglas Created a Tremendous Legacy in the Plants That He Intro (P Coulteri) Pines
The American lIorHcullural Sociely inviles you Io Celehrate tbe American Gardener al our 1999 Annual Conference Roston" Massachusetts June 9 - June 12~ 1999 Celebrate Ute accompHsbenls of American gardeners in Ute hlsloric "Cay Upon lhe 1Iill." Join wah avid gardeners from. across Ute counlrg lo learn new ideas for gardening excellence. Attend informa-Hve ledures and demonslraHons by naHonally-known garden experts. Tour lhe greal public and privale gardens in and around Roslon, including Ute Arnold Arborelum and Garden in Ute Woods. Meet lhe winners of AIlS's 1999 naHonJ awards for excellence in horHcullure. @ tor more informaHon, call1he conference regislrar al (800) 777-7931 ext 10. co n t e n t s Volume 78, Number 1 • '.I " Commentary 4 Hellebores 22 Members' Forum 5 by C. Colston Burrell Staghorn fern) ethical plant collecting) orchids. These early-blooming pennnials are riding the crest of a wave ofpopularity) and hybridizers are News from AHS 7 busy working to meet the demand. Oklahoma Horticultural Society) Richard Lighty) Robert E. Lyons) Grecian foxglove. David Douglas 30 by Susan Davis Price Focus 9 Many familiar plants in cultivation today New plants for 1999. are improved selections of North American species Offshoots 14 found by this 19th-century Scottish expLorer. Waiting for spring in Vermont. Bold Plants 37 Gardeners Information Service 15 by Pam Baggett Houseplants) transplanting a ginkgo tree) Incorporating a few plants with height) imposing starting trees from seed) propagating grape vines. foliage) or striking blossoms can make a dramatic difference in any landscape design. Mail-Order Explorer 16 Heirloom flowers and vegetables. -
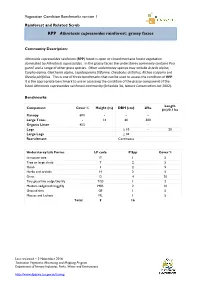
RPP Athrotaxis Cupressoides Rainforest: Grassy Facies
Vegetation Condition Benchmarks version 1 Rainforest and Related Scrub RPP Athrotaxis cupressoides rainforest: grassy facies Community Description: Athrotaxis cupressoides rainforest (RPP) forest is open or closed montane forest vegetation dominated by Athrotaxis cupressoides. In the grassy facies the understorey commonly contains Poa gunnii and a range of other grass species. Other understorey species may include Astelia alpina, Carpha alpina, Gleichenia alpina, Lepidosperma filiforme, Oreobolus distichus, Richea scoparia and Olearia pinifolius. This is one of three benchmarks that can be used to assess the condition of RPP. It is the appropriate benchmark to use in assessing the condition of the grassy component of the listed Athrotaxis cupressoides rainforest community (Schedule 3A, Nature Conservation Act 2002). Benchmarks: Length Component Cover % Height (m) DBH (cm) #/ha (m)/0.1 ha Canopy 60% - - - Large Trees - 14 60 300 Organic Litter 45% - Logs ≥ 10 - 20 Large Logs ≥ 30 Recruitment Continuous Understorey Life Forms LF code # Spp Cover % Immature tree IT 1 5 Tree or large shrub T 2 5 Shrub S 2 5 Herbs and orchids H 2 5 Grass G 4 20 Tiny grass/tiny sedge/tiny lily TGS 1 2 Medium sedge/rush/sagg/lily MSR 2 10 Ground fern GF 1 5 Mosses and Lichens ML 1 5 Total 9 16 Last reviewed – 2 November 2016 Tasmanian Vegetation Monitoring and Mapping Program Department of Primary Industries, Parks, Water and Environment http://www.dpipwe.tas.gov.au/tasveg RPP Athrotaxis cupressoides rainforest: grassy facies Species lists: Canopy Tree Species Common Name Notes Athrotaxis cupressoides pencil pine Cradle Mountain area; some Athrotaxis selaginoides king billy pine closed canopy forests Cradle Mountain area; some Nothofagus cunninghamii myrtle beech closed canopy forests Typical Understorey Species * Common Name LF Code Nothofagus cunninghamii myrtle beech T Phyllocladus aspleniifolius celery top pine T Olearia pinifolia prickly daisybush S Richea species richea S Poa gunnii gunns snowgrass G Rytidosperma spp. -

Trees, Shrubs, and Perennials That Intrigue Me (Gymnosperms First
Big-picture, evolutionary view of trees and shrubs (and a few of my favorite herbaceous perennials), ver. 2007-11-04 Descriptions of the trees and shrubs taken (stolen!!!) from online sources, from my own observations in and around Greenwood Lake, NY, and from these books: • Dirr’s Hardy Trees and Shrubs, Michael A. Dirr, Timber Press, © 1997 • Trees of North America (Golden field guide), C. Frank Brockman, St. Martin’s Press, © 2001 • Smithsonian Handbooks, Trees, Allen J. Coombes, Dorling Kindersley, © 2002 • Native Trees for North American Landscapes, Guy Sternberg with Jim Wilson, Timber Press, © 2004 • Complete Trees, Shrubs, and Hedges, Jacqueline Hériteau, © 2006 They are generally listed from most ancient to most recently evolved. (I’m not sure if this is true for the rosids and asterids, starting on page 30. I just listed them in the same order as Angiosperm Phylogeny Group II.) This document started out as my personal landscaping plan and morphed into something almost unwieldy and phantasmagorical. Key to symbols and colored text: Checkboxes indicate species and/or cultivars that I want. Checkmarks indicate those that I have (or that one of my neighbors has). Text in blue indicates shrub or hedge. (Unfinished task – there is no text in blue other than this text right here.) Text in red indicates that the species or cultivar is undesirable: • Out of range climatically (either wrong zone, or won’t do well because of differences in moisture or seasons, even though it is in the “right” zone). • Will grow too tall or wide and simply won’t fit well on my property.