The Epigenome in Pluripotency and Differentiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Altered DNA Methylation Pattern Characterizes the Peripheral Immune Cells of Patients with Autoimmune Hepatitis
medRxiv preprint doi: https://doi.org/10.1101/2021.07.01.21259836; this version posted July 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Altered DNA methylation pattern characterizes the peripheral immune cells of patients with autoimmune hepatitis Kalliopi Zachou1,2*, Pinelopi Arvaniti1*, Aggeliki Lyberopoulou1, Eirini Sevdali3, Matthaios Speletas3, Maria Ioannou4, George K. Koukoulis4, Yves Renaudineau5,6**, George N. Dalekos1,2** *These authors contributed equally to this work **These authors contributed equally to this work as senior authors 1Department of Medicine and Research Laboratory of Internal Medicine, National Expertise Center of Greece in Autoimmune Liver Diseases, General University Hospital of Larissa, 41110 Larissa, Greece; 2Institute of Internal Medicine and Hepatology, 41447 Larissa, Greece; 3Department of Immunology and Histocompatibility, Faculty of Medicine, School of Health Sciences, University of Thessaly, Larissa, Greece; 4Department of Pathology, Faculty of Medicine, School of Health Sciences, University of Thessaly, Larissa, Greece; 5INSERN U1291, CNR U5051, University Toulouse III, Toulouse Institute for infectious and inflammatory diseases, Toulouse, France; 6Department of Immunology, Purpan University Hospital Toulouse, Toulouse, France All correspondence and reprint requests to: George N. Dalekos, MD, PhD Professor of Medicine Head, Department of Medicine and Research Laboratory of Internal Medicine National Expertise Center of Greece in Autoimmune Liver Diseases General University Hospital of Larissa, 41110 Larissa, Greece Tel: ++30-241-350-2285; Fax: ++30-241-350-1557 e-mail: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. -

ZFP57 Gene ZFP57 Zinc Finger Protein
ZFP57 gene ZFP57 zinc finger protein Normal Function The protein produced from the ZFP57 gene is a member of a family called zinc finger proteins, which are involved in many cellular functions. Zinc finger proteins each contain one or more short regions called zinc finger domains. These regions include a specific pattern of protein building blocks (amino acids) and one or more charged atoms of zinc ( zinc ions). Zinc finger proteins attach (bind) primarily to DNA. In most cases, these proteins attach to regions near certain genes and turn the genes on and off as needed. Proteins that bind to DNA and regulate the activity of particular genes are known as transcription factors. Some zinc finger proteins can also bind to other molecules, including RNA (a chemical cousin of DNA) and proteins. The ZFP57 protein is involved in the regulation of other genes by the addition of methyl groups, consisting of one carbon atom and three hydrogen atoms (methylation). Methylation is important in many cellular functions. These include determining whether the instructions in a particular segment of DNA are carried out or suppressed (gene silencing), regulating reactions involving proteins and lipids, and controlling the processing of chemicals that relay signals in the nervous system (neurotransmitters). Health Conditions Related to Genetic Changes 6q24-related transient neonatal diabetes mellitus Mutations in the ZFP57 gene cause about 10 percent of cases of 6q24-related transient neonatal diabetes mellitus, a type of diabetes that occurs in infants. This disorder results from the overactivity (overexpression) of certain genes in a region of the long (q) arm of chromosome 6 called 6q24. -

WO 2019/089592 Al 09 May 2019 (09.05.2019) W 1 P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization III International Bureau (10) International Publication Number (43) International Publication Date WO 2019/089592 Al 09 May 2019 (09.05.2019) W 1 P O PCT (51) International Patent Classification: (72) Inventors; and A61K 35/12 (2015.01) C07K 16/30 (2006.01) (71) Applicants: JAN, Max [US/US]; c/o The Broad Institute, A61K 5/7 7 (2015.01) C07K 14/705 (2006.01) Inc., 45 1 Main Street, Cambridge, Massachusetts 02142 (US). SIEVERS, Quinlan [US/US]; c/o The Broad In¬ (21) International Application Number: stitute, Inc., 451 Main Street, Cambridge, Massachusetts PCT/US2018/058210 02142 (US). EBERT, Benjamin [US/US]; c/o The Broad (22) International Filing Date: Institute, Inc., 451 Main Street, Cambridge, Massachusetts 30 October 2018 (30. 10.2018) 02142 (US). MAUS, Marcela [US/US]; c/o The Broad In¬ stitute, Inc., 451 Main Street, Cambridge, Massachusetts (25) Filing Language: English 02142 (US). (26) Publication Language: English (74) Agent: COWLES, Christopher R. et al; Burns & Levin- (30) Priority Data: son, LLP, 125 Summer Street, Boston, Massachusetts 62/579,454 3 1 October 2017 (3 1. 10.2017) U S 021 10 (US). 62/633,725 22 February 2018 (22.02.2018) U S (81) Designated States (unless otherwise indicated, for every (71) Applicants: THE GENERAL HOSPITAL CORPO¬ kind of national protection available): AE, AG, AL, AM, RATION [US/US]; 55 Fruit Street, Boston, Massachu¬ AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, setts 021 14 (US) BRIGHAM & WOMEN'S HOSPITAL CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, [US/US]; 75 Francis Street, Boston, Massachusetts 021 15 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (US) PRESIDENT AND FELLOWS OF HARVARD HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, COLLEGE [US/US]; 17 Quincy Street, Cambridge, Mass¬ KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, achusetts 02138 (US). -
Supplementary Items
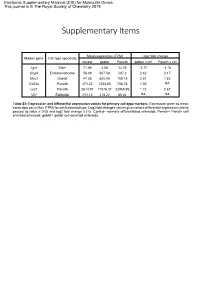
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2019 Supplementary Items Mean expression (TPM) Log2 fold change Marker gene Cell type specificity control goblet Paneth goblet v ctrl Paneth v ctrl Lgr5 Stem 71.88 4.08 12.76 -2.77 -1.16 ChgA Enteroendocrine 56.49 507.58 287.2 2.42 2.17 Muc2 Goblet 47.26 625.05 185.18 2.81 1.83 Cd24a Paneth 471.23 1034.85 708.78 1.05 NA Lyz1 Paneth 2613.91 11576.37 22065.95 1.72 2.67 Vil1 Epithelial 211.13 219.22 89.02 NA NA Table S2: Expression and differential expression values for primary cell-type markers. Expression given as mean transcripts per million (TPM) for each enteroid type. Log2 fold changes values given where differential expression criteria passed (q value ≤ 0.05 and log2 fold change ≥ |1|). Control- normally differentiated enteroids; Paneth= Paneth cell enriched enteroids; goblet= goblet cell enriched enteroids. Paneth Paneth Goblet Goblet Gene Type Gene Name Ensembl ID DEG lfc DEG fdr DEG lfc DEG fdr AMP Ang4 ENSMUSG00000060615 4.64 8.61E-36 2.45 8.75E-13 AMP Defa17 ENSMUSG00000060208 4.02 6.40E-46 2.95 5.56E-31 AMP Defa2 ENSMUSG00000096295 4.58 1.70E-18 NA NA AMP Defa20 ENSMUSG00000095066 4.40 1.19E-17 NA NA AMP Defa21 ENSMUSG00000074447 5.18 2.22E-22 NA NA AMP Defa22 ENSMUSG00000074443 5.40 3.09E-20 NA NA AMP Defa23 ENSMUSG00000074446 3.69 1.94E-12 NA NA AMP Defa24 ENSMUSG00000064213 3.91 8.51E-38 3.24 1.92E-32 AMP Defa26 ENSMUSG00000060070 3.02 1.86E-39 2.22 8.61E-27 AMP Defa28 ENSMUSG00000074434 2.84 3.21E-17 1.66 1.20E-07 -

Stem Cell 2015;6(2) 56
Stem Cell 2015;6(2) http://www.sciencepub.net/stem Stem Cell and Transcript Research Literatures Ma Hongbao 1, Margaret Young 2, Yang Yan 1 1 Brookdale Hospital, Brooklyn, New York 11212, USA; 2 Cambridge, MA 02138, USA [email protected] Abstract: The stem cell is the origin of an organism’s life that has the potential to develop into many different types of cells in life bodies. In many tissues stem cells serve as a sort of internal repair system, dividing essentially without limit to replenish other cells as long as the person or animal is still alive. When a stem cell divides, each new cell has the potential either to remain a stem cell or become another type of cell with a more specialized function, such as a red blood cell or a brain cell. This article introduces recent research reports as references in the stem cell and transcript related studies. [Ma H, Young M, Yang Y. Stem Cell and Transcript Research Literatures. Stem Cell. 2015;6(2):56-72] (ISSN 1545-4570). http://www.sciencepub.net/stem. 8 Key words: stem cell; transcript; life; research; literature 1. Introduction REST (322) target genes. The majority of these The stem cell is the origin of an organism’s life CoREST targets do not contain known RE1 motifs. that has the potential to develop into many different Notably, these CoREST target genes do play important types of cells in life bodies. In many tissues stem cells roles in pluripotency networks, in modulating NSC serve as a sort of internal repair system, dividing identity and fate decisions and in epigenetic processes essentially without limit to replenish other cells as previously associated with both REST and CoREST. -

WO 2012/054896 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number ι (43) International Publication Date ¾ ί t 2 6 April 2012 (26.04.2012) WO 2012/054896 Al (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, C12N 5/00 (2006.01) C12N 15/00 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, C12N 5/02 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (21) International Application Number: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, PCT/US201 1/057387 ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (22) International Filing Date: NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, 2 1 October 201 1 (21 .10.201 1) RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (25) Filing Language: English ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/406,064 22 October 2010 (22.10.2010) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, 61/415,244 18 November 2010 (18.1 1.2010) US UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, (71) Applicant (for all designated States except US): BIO- DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, TIME INC. -

Regulatory Network Analysis of Paneth Cell and Goblet Cell Enriched Gut Organoids Using Transcriptomics Approaches
bioRxiv preprint doi: https://doi.org/10.1101/575845; this version posted October 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Regulatory network analysis of Paneth cell and goblet cell enriched gut organoids using transcriptomics approaches Treveil A1,2*, Sudhakar P1,3*, Matthews Z J4*, Wrzesinski T1, Jones E J1,2, Brooks J1,2,4,5, Olbei M1,2, Hautefort I1, Hall L J2, Carding S R2,4, Mayer U6, Powell P P4, Wileman T2,4, Di Palma F1, Haerty W1,#, Korcsmáros T1,2# 1 Earlham Institute, Norwich Research Park, Norwich, Norfolk, NR4 7UZ, UK 2 Quadram Institute, Norwich Research Park, Norwich, Norfolk, NR4 7UA, UK 3 Department of Chronic Diseases, Metabolism and Ageing, KU Leuven, Belgium 4 Norwich Medical School, University of East Anglia, Norwich, Norfolk, NR4 7TJ, UK 5 Department of Gastroenterology, Norfolk and Norwich University Hospitals, Norwich, UK 6 School of Biological Sciences, University of East Anglia, Norwich, Norfolk, NR4 7TJ, UK * Equal contributions # Joint corresponding authors Corresponding author contact details: Dr Tamas Korcsmaros ORCID id : https://orcid.org/0000-0003-1717-996X [email protected] Phone : 0044-1603450961 Fax : 0044-1603450021 Address: Earlham Institute, Norwich Research Park, Norwich, NR4 7UZ, UK 1 bioRxiv preprint doi: https://doi.org/10.1101/575845; this version posted October 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Identification of Biomarkers, Pathways and Potential Therapeutic Targets for Heart Failure Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.05.455244; this version posted August 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of biomarkers, pathways and potential therapeutic targets for heart failure using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.08.05.455244; this version posted August 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Heart failure (HF) is a complex cardiovascular diseases associated with high mortality. To discover key molecular changes in HF, we analyzed next-generation sequencing (NGS) data of HF. In this investigation, differentially expressed genes (DEGs) were analyzed using limma in R package from GSE161472 of the Gene Expression Omnibus (GEO). Then, gene enrichment analysis, protein-protein interaction (PPI) network, miRNA-hub gene regulatory network and TF-hub gene regulatory network construction, and topological analysis were performed on the DEGs by the Gene Ontology (GO), REACTOME pathway, STRING, HiPPIE, miRNet, NetworkAnalyst and Cytoscape. Finally, we performed receiver operating characteristic curve (ROC) analysis of hub genes. A total of 930 DEGs 9464 up regulated genes and 466 down regulated genes) were identified in HF. -

Chromatin Signatures of Pluripotent Cell Lines
LETTERS Chromatin signatures of pluripotent cell lines Véronique Azuara 1,2,7, Pascale Perry1, Stephan Sauer1, Mikhail Spivakov1, Helle F. Jørgensen1, Rosalind M. John1,3, Mina Gouti1,4, Miguel Casanova5, Gary Warnes6, Matthias Merkenschlager1 and Amanda G. Fisher1,7 Epigenetic genome modifications are thought to be important (HSCs). Early replication during S phase is widely considered a feature for specifying the lineage and developmental stage of of gene-rich, transcriptionally active regions of the genome and cor- cells within a multicellular organism. Here, we show that relates with accessible chromatin carrying acetylated histones, whereas the epigenetic profile of pluripotent embryonic stem cells late replication is characteristic of constitutive heterochromatin and (ES) is distinct from that of embryonic carcinoma cells, some facultative heterochromatin15–17. The timing of DNA replication haematopoietic stem cells (HSC) and their differentiated of a panel of transcriptional regulatory genes (which are important for progeny. Silent, lineage-specific genes replicated earlier early development or for the specification of neural, haematopoietic in pluripotent cells than in tissue-specific stem cells or or muscle lineages) was compared in pluripotent ES cells, multipotent differentiated cells and had unexpectedly high levels of haematopoietic cell lines and unipotent T lymphocytes (Fig. 1 and see acetylated H3K9 and methylated H3K4. Unusually, in ES Supplementary Information, Fig. S1). Replication timing was assessed cells these markers