Characterizing Iqgap1-C Domain Binding to Phosphoinositides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dnmt3a Restrains Mast Cell Inflammatory Responses PNAS PLUS
Dnmt3a restrains mast cell inflammatory responses PNAS PLUS Cristina Leonia,b, Sara Montagnera, Andrea Rinaldic, Francesco Bertonic,d, Sara Pollettie, Chiara Balestrierie, and Silvia Monticellia,1 aInstitute for Research in Biomedicine, Università della Svizzera italiana (USI), 6500 Bellinzona, Switzerland; bGraduate School for Cellular and Biomedical Sciences, University of Bern, 3012 Bern, Switzerland; cLymphoma and Genomics Research Program, Institute of Oncology Research (IOR), USI, 6500 Bellinzona, Switzerland; dOncology Institute of Southern Switzerland (IOSI), 6500 Bellinzona, Switzerland; and eDepartment of Experimental Oncology, European Institute of Oncology (IEO), 20139 Milan, Italy Edited by Anjana Rao, Sanford Consortium for Regenerative Medicine and La Jolla Institute for Allergy and Immunology, La Jolla, CA, and approved January 10, 2017 (received for review October 3, 2016) DNA methylation and specifically the DNA methyltransferase mast cells (which are key effector cells in asthmatic and allergic enzyme DNMT3A are involved in the pathogenesis of a variety responses), a recent survey compared atopic and asthmatic pa- of hematological diseases and in regulating the function of immune tients with healthy controls and identified 81 differentially meth- cells. Although altered DNA methylation patterns and mutations in ylated regions (15); the hypomethylated regions included genes DNMT3A correlate with mast cell proliferative disorders in humans, such as IL13, which is not only crucial in asthma pathogenesis but the role of DNA methylation in mast cell biology is not understood. By is also expressed at high levels by mast cells (16). Finally, high- using mast cells lacking Dnmt3a, we found that this enzyme is in- lighting the potential relevance of understanding the role of DNA volved in restraining mast cell responses to acute and chronic stimuli, methylation in mast cell biology, altered DNA methylation pat- both in vitro and in vivo. -

Role of IQGAP1 in Carcinogenesis
cancers Review Role of IQGAP1 in Carcinogenesis Tao Wei and Paul F. Lambert * McArdle Laboratory for Cancer Research, Department of Oncology, University of Wisconsin School of Medicine and Public Health, Madison, WI 53705, USA; [email protected] * Correspondence: [email protected] Simple Summary: IQ motif-containing GTPase-activating protein 1 (IQGAP1) is a signal scaffolding protein that regulates a range of cellular activities by facilitating signal transduction in cells. IQGAP1 is involved in many cancer-related activities, such as proliferation, apoptosis, migration, invasion and metastases. In this article, we review the different pathways regulated by IQGAP1 during cancer development, and the role of IQGAP1 in different types of cancer, including cancers of the head and neck, breast, pancreas, liver, colorectal, stomach, and ovary. We also discuss IQGAP10s regulation of the immune system, which is of importance to cancer progression. This review highlights the significant roles of IQGAP1 in cancer and provides a rationale for pursuing IQGAP1 as a drug target for developing novel cancer therapies. Abstract: Scaffolding proteins can play important roles in cell signaling transduction. IQ motif- containing GTPase-activating protein 1 (IQGAP1) influences many cellular activities by scaffolding multiple key signaling pathways, including ones involved in carcinogenesis. Two decades of studies provide evidence that IQGAP1 plays an essential role in promoting cancer development. IQGAP1 is overexpressed in many types of cancer, and its overexpression in cancer is associated with lower survival of the cancer patient. Here, we provide a comprehensive review of the literature regarding the oncogenic roles of IQGAP1. We start by describing the major cancer-related signaling pathways Citation: Wei, T.; Lambert, P.F. -

A Study of Calponin's Role in Secretion
WORCESTER POLYTECHNIC INSTITUTE A Study of Calponin’s Role in Secretion The Proposed Model and the Experimental Design Christopher T. Rollins 4/30/2009 A Major Qualifying Project Report Submitted to the Faculty of the WORCESTER POLYTECHNIC INSTITUTE in partial fulfillment of the requirements for the Degree of Bachelor of Science in Biology and Biotechnology Approved: Jill Rulfs – Advisor Keywords: Actin, Cytoskeleton, Calponin, CaP, CN , Secretion , Exocytosis 1 A Study of Calponin’s R ole in Secretion Table of Contents Abstract .................................................................................................................................................. 2 Background ............................................................................................................................................. 2 Introduction ............................................................................................................................................ 6 Secretion Study – Model, Hypothesis, and Experiments ................................................................... 7 Materials and Methods ........................................................................................................................ 11 Amplification of pET, pCMV-HA, and GFP-Calponin Vectors ........................................................... 11 Creation of DH5α and XL-10 Gold Calcium Competent Cells ........................................................... 11 Transformation of Calcium Competent Cells .................................................................................. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Views for Entrez
BASIC RESEARCH www.jasn.org Phosphoproteomic Analysis Reveals Regulatory Mechanisms at the Kidney Filtration Barrier †‡ †| Markus M. Rinschen,* Xiongwu Wu,§ Tim König, Trairak Pisitkun,¶ Henning Hagmann,* † † † Caroline Pahmeyer,* Tobias Lamkemeyer, Priyanka Kohli,* Nicole Schnell, †‡ †† ‡‡ Bernhard Schermer,* Stuart Dryer,** Bernard R. Brooks,§ Pedro Beltrao, †‡ Marcus Krueger,§§ Paul T. Brinkkoetter,* and Thomas Benzing* *Department of Internal Medicine II, Center for Molecular Medicine, †Cologne Excellence Cluster on Cellular Stress | Responses in Aging-Associated Diseases, ‡Systems Biology of Ageing Cologne, Institute for Genetics, University of Cologne, Cologne, Germany; §Laboratory of Computational Biology, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland; ¶Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand; **Department of Biology and Biochemistry, University of Houston, Houston, Texas; ††Division of Nephrology, Baylor College of Medicine, Houston, Texas; ‡‡European Molecular Biology Laboratory–European Bioinformatics Institute, Hinxton, Cambridge, United Kingdom; and §§Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany ABSTRACT Diseases of the kidney filtration barrier are a leading cause of ESRD. Most disorders affect the podocytes, polarized cells with a limited capacity for self-renewal that require tightly controlled signaling to maintain their integrity, viability, and function. Here, we provide an atlas of in vivo phosphorylated, glomerulus- expressed -

Zimmer Cell Calcium 2013 Mammalian S100 Evolution.Pdf
Cell Calcium 53 (2013) 170–179 Contents lists available at SciVerse ScienceDirect Cell Calcium jo urnal homepage: www.elsevier.com/locate/ceca Evolution of the S100 family of calcium sensor proteins a,∗ b b,1 b Danna B. Zimmer , Jeannine O. Eubanks , Dhivya Ramakrishnan , Michael F. Criscitiello a Center for Biomolecular Therapeutics and Department of Biochemistry & Molecular Biology, University of Maryland School of Medicine, 108 North Greene Street, Baltimore, MD 20102, United States b Comparative Immunogenetics Laboratory, Department of Veterinary Pathobiology, College of Veterinary Medicine & Biomedical Sciences, Texas A&M University, College Station, TX 77843-4467, United States a r t i c l e i n f o a b s t r a c t 2+ Article history: The S100s are a large group of Ca sensors found exclusively in vertebrates. Transcriptomic and genomic Received 4 October 2012 data from the major radiations of mammals were used to derive the evolution of the mammalian Received in revised form 1 November 2012 S100s genes. In human and mouse, S100s and S100 fused-type proteins are in a separate clade from Accepted 3 November 2012 2+ other Ca sensor proteins, indicating that an ancient bifurcation between these two gene lineages Available online 14 December 2012 has occurred. Furthermore, the five genomic loci containing S100 genes have remained largely intact during the past 165 million years since the shared ancestor of egg-laying and placental mammals. Keywords: Nonetheless, interesting births and deaths of S100 genes have occurred during mammalian evolution. Mammals The S100A7 loci exhibited the most plasticity and phylogenetic analyses clarified relationships between Phylogenetic analyses the S100A7 proteins encoded in the various mammalian genomes. -

Hyperactivity Disorder
Molecular Psychiatry (2011) 16, 491–503 & 2011 Macmillan Publishers Limited All rights reserved 1359-4184/11 www.nature.com/mp ORIGINAL ARTICLE Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree K-P Lesch1,6, S Selch1,6, TJ Renner2,6, C Jacob1, TT Nguyen3, T Hahn1, M Romanos2, S Walitza2, S Shoichet4, A Dempfle3, M Heine1, A Boreatti-Hu¨mmer1, J Romanos1, S Gross-Lesch1, H Zerlaut3, T Wultsch1, S Heinzel1, M Fassnacht5, A Fallgatter1, B Allolio5, H Scha¨fer4, A Warnke2, A Reif1, H-H Ropers4 and R Ullmann4 1ADHD Clinical Research Network, Unit for Molecular Psychiatry, Laboratory of Translational Neuroscience, Department of Psychiatry, Psychosomatics and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany; 2ADHD Clinical Research Network, Department of Child and Adolescent Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany; 3Institute of Medical Biometry and Epidemiology, University of Marburg, Marburg, Germany; 4Max Planck Institute for Molecular Genetics, Berlin, Germany and 5Department of Endocrinology and Diabetes, Internal Medicine I, University of Wuerzburg, Wuerzburg, Germany Attention-deficit/hyperactivity disorder (ADHD) is a common, highly heritable neurodevelop- mental syndrome characterized by hyperactivity, inattention and increased impulsivity. To detect micro-deletions and micro-duplications that may have a role in the pathogenesis of ADHD, we carried out a genome-wide screen for copy number variations (CNVs) in a cohort of 99 children and adolescents with severe ADHD. Using high-resolution array comparative genomic hybridization (aCGH), a total of 17 potentially syndrome-associated CNVs were identified. The aberrations comprise 4 deletions and 13 duplications with approximate sizes ranging from 110 kb to 3 Mb. -

Endogenous IQGAP1 and IQGAP3 Do Not Functionally Interact with Ras Chase J
www.nature.com/scientificreports OPEN Endogenous IQGAP1 and IQGAP3 do not functionally interact with Ras Chase J. Morgan, Andrew C. Hedman, Zhigang Li & David B. Sacks The Ras family of small GTPases modulates numerous essential processes. Activating Ras mutations Received: 18 January 2019 result in hyper-activation of selected signaling cascades, which leads to human diseases. The Accepted: 28 June 2019 high frequency of Ras mutations in human malignant neoplasms has led to Ras being a desirable Published: xx xx xxxx chemotherapeutic target. The IQGAP family of scafold proteins binds to and regulates multiple signaling molecules, including the Rho family GTPases Rac1 and Cdc42. There are conficting data in the published literature regarding interactions between IQGAP and Ras proteins. Initial reports showed no binding, but subsequent studies claim associations of IQGAP1 and IQGAP3 with K-Ras and H-Ras, respectively. Therefore, we set out to resolve this controversy. Here we demonstrate that neither endogenous IQGAP1 nor endogenous IQGAP3 binds to the major Ras isoforms, namely H-, K-, and N-Ras. Importantly, Ras activation by epidermal growth factor is not altered when IQGAP1 or IQGAP3 proteins are depleted from cells. These data strongly suggest that IQGAP proteins are not functional interactors of H-, K-, or N-Ras and challenge the rationale for targeting the interaction of Ras with IQGAP for the development of therapeutic agents. Te Ras superfamily of small GTPases contains >150 proteins in humans1. Tese are grouped into 5 families based on sequence and functional diferences. Te Ras family, which includes the isoforms H-, K- and N-Ras1, controls cell proliferation, survival and cell diferentiation. -

Contractility Kits Promote Assembly of the Mechanoresponsive
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs226704. doi:10.1242/jcs.226704 RESEARCH ARTICLE Contractility kits promote assembly of the mechanoresponsive cytoskeletal network Priyanka Kothari1, Vasudha Srivastava1,2,*, Vasudha Aggarwal3,‡, Irina Tchernyshyov4, Jennifer E. Van Eyk4, Taekjip Ha3,5,6,7 and Douglas N. Robinson1,2,8,9,§ ABSTRACT modulate their behavior in response to internal and external cues, Cellular contractility is governed by a control system of proteins that and regulatory proteins integrate those cues through feedback loops integrates internal and external cues to drive diverse shape change to further tune the response of the cell. Cellular contractility is a processes. This contractility controller includes myosin II motors, actin prime example of such a control system, as it is driven by molecular crosslinkers and protein scaffolds, which exhibit robust and cooperative machinery that is spatially and temporally regulated by feedback mechanoaccumulation. However, the biochemical interactions and loops (Effler et al., 2006; Ren et al., 2009; Luo et al., 2012, 2013; feedback mechanisms that drive the controller remain unknown. Here, Schiffhauer et al., 2016) (Fig. 1A). For example, during cell we use a proteomics approach to identify direct interactors of two key migration, chemical cues drive actin polymerization at the leading nodes of the contractility controller in the social amoeba Dictyostelium edge, resulting in tension in the cortical network that causes the discoideum: the actin crosslinker cortexillin I and the scaffolding protein contractile machinery to accumulate at the lagging edge, thereby IQGAP2. We highlight several unexpected proteins that suggest allowing the cell to move forward (Lee et al., 2010). -

UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent Permalink https://escholarship.org/uc/item/7m04f0b0 Author Buchholz, Kyle Stephen Publication Date 2016 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Kyle Stephen Buchholz Committee in Charge: Professor Jeffrey Omens, Chair Professor Andrew McCulloch, Co-Chair Professor Ju Chen Professor Karen Christman Professor Robert Ross Professor Alexander Zambon 2016 Copyright Kyle Stephen Buchholz, 2016 All rights reserved Signature Page The Dissertation of Kyle Stephen Buchholz is approved and it is acceptable in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California, San Diego 2016 iii Dedication To my beautiful wife, Rhia. iv Table of Contents Signature Page ................................................................................................................... iii Dedication .......................................................................................................................... iv Table of Contents ................................................................................................................ v List of Figures ................................................................................................................... -

13074 IQGAP2 (D1X8U) Rabbit Mab
Revision 1 C 0 2 - t IQGAP2 (D1X8U) Rabbit mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] 4 Support: 877-678-TECH (8324) 7 0 Web: [email protected] 3 www.cellsignal.com 1 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB H Endogenous 180 Rabbit IgG Q13576 10788 Product Usage Information 7. Jin, S.H. et al. (2008) Int J Cancer 122, 1040-6. 8. Liu, Z. et al. (2010) Clin Cancer Res 16, 6009-18. Application Dilution 9. Wang, S. et al. (2007) J Cell Sci 120, 567-77. 10. Schmidt, V.A. et al. (2003) Blood 101, 3021-8. Western Blotting 1:1000 11. Jin, S.H. et al. (2008) Int J Cancer 122, 1040-6. 12. Nojima, H. et al. (2008) Nat Cell Biol 10, 971-8. Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity IQGAP2 (D1X8U) Rabbit mAb recognizes endogenous levels of total IQGAP2 protein. Species Reactivity: Human Source / Purification Monoclonal antibody is produced by immunizing animals with a synthetic peptide corresponding to residues near the carboxy terminus of human IQGAP2 protein. Background IQGAPs are scaffolding proteins involved in mediating cytoskeletal function. They contain multiple protein interaction domains and bind to a growing number of molecules including actin, myosin light chain, calmodulin, E-cadherin, and β-catenin (reviewed in 1). -
Supplementary Figures S1-S3
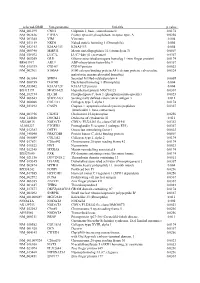
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089