From Fossils to Phylogenies Part 1: Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Megafauna Extinction, Tree Species Range Reduction, and Carbon Storage in Amazonian Forests
Ecography 39: 194–203, 2016 doi: 10.1111/ecog.01587 © 2015 The Authors. Ecography © 2015 Nordic Society Oikos Subject Editor: Yadvinder Mahli. Editor-in-Chief: Nathan J. Sanders. Accepted 27 September 2015 Megafauna extinction, tree species range reduction, and carbon storage in Amazonian forests Christopher E. Doughty, Adam Wolf, Naia Morueta-Holme, Peter M. Jørgensen, Brody Sandel, Cyrille Violle, Brad Boyle, Nathan J. B. Kraft, Robert K. Peet, Brian J. Enquist, Jens-Christian Svenning, Stephen Blake and Mauro Galetti C. E. Doughty ([email protected]), Environmental Change Inst., School of Geography and the Environment, Univ. of Oxford, South Parks Road, Oxford, OX1 3QY, UK. – A. Wolf, Dept of Ecology and Evolutionary Biology, Princeton Univ., Princeton, NJ 08544, USA. – N. Morueta-Holme, Dept of Integrative Biology, Univ. of California – Berkeley, CA 94720, USA. – B. Sandel, J.-C. Svenning and NM-H, Section for Ecoinformatics and Biodiversity, Dept of Bioscience, Aarhus Univ., Ny Munkegade 114, DK-8000 Aarhus C, Denmark. – P. M. Jørgensen, Missouri Botanical Garden, PO Box 299, St Louis, MO 63166-0299, USA. – C. Violle, CEFE UMR 5175, CNRS – Univ. de Montpellier – Univ. Paul-Valéry Montpellier – EPHE – 1919 route de Mende, FR-34293 Montpellier Cedex 5, France. – B. Boyle and B. J. Enquist, Dept of Ecology and Evolutionary Biology, Univ. of Arizona, Tucson, AZ 85721, USA. – N. J. B. Kraft, Dept of Biology, Univ. of Maryland, College Park, MD 20742, USA. BJE also at: The Santa Fe inst., 1399 Hyde Park Road, Santa Fe, NM 87501, USA. – R. K. Peet, Dept of Biology, Univ. of North Carolina, Chapel Hill, NC 27599-3280, USA. -

Pleistocene Mammals and Paleoecology of the Western Amazon
PLEISTOCENE MAMMALS AND PALEOECOLOGY OF THE WESTERN AMAZON By ALCEU RANCY A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 1991 . To Cleusa, Bianca, Tiago, Thomas, and Nono Saul (Pistolin de Oro) . ACKNOWLEDGMENTS This work received strong support from John Eisenberg (chairman) and David Webb, both naturalists, humanists, and educators. Both were of special value, contributing more than the normal duties as members of my committee. Bruce MacFadden provided valuable insights at several periods of uncertainty. Ronald Labisky and Kent Redford also provided support and encouragement. My field work in the western Amazon was supported by several grants from the Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) , and the Universidade Federal do Acre (UFAC) , Brazil. I also benefitted from grants awarded to Ken Campbell and Carl Frailey from the National Science Foundation (NSF) I thank Daryl Paul Domning, Jean Bocquentin Villanueva, Jonas Pereira de Souza Filho, Ken Campbell, Jose Carlos Rodrigues dos Santos, David Webb, Jorge Ferigolo, Carl Frailey, Ernesto Lavina, Michael Stokes, Marcondes Costa, and Ricardo Negri for sharing with me fruitful and adventurous field trips along the Amazonian rivers. The CNPq and the Universidade Federal do Acre, supported my visit to the. following institutions (and colleagues) to examine their vertebrate collections: iii . ; ; Universidade do Amazonas, Manaus -

Searchable PDF (No Class Notes) (9.906Mb)
The paper used in this publication is manufactured from 100%-recycled fiber of which 50% is recovered from Post Consumer sources. This product is made exclusively for New Leaf Paper in San Francisco, California by Plainwell Paper Company and was offered to Kalamazoo College with their permis Jllege learning experience takes this young member of sion. New Leaf Paper is nationally known for their active role in the design and marketing of paper products that II which people and communities the power of that are entirely recycled and recyclable. Two members of the Kalamazoo College Jirectly, as she uses it to build her future. Class of 1963 had active roles in the manufacture of these items. rndations that support Kalamazoo College. The others 1e. All are vital. Your Annual Fund gift makes the ~e possible for young people like her. Most annual fund gifts are applied to student scholarships and program or facility improvements. Annual fund gifts are unrestricted, which confers upon the College the opportunity to use them in ways to best support the K-Plan. And the degree of Annual Fund participation unlocks additional gifts from corporations and foundations. Alumni participation is one of the first facts requested by grant officers when they consider major funding proposals from the College. So thank you again, alumni and friends. For the second consecutive year the Annual Fund reached and exceeded its goal. It will help enlighten futures. KALAMAZOO COLLEGE A~fiutd 1 9 9 8 - 1 9 9 9 Time will tell where her Kalamazoo College learning experience takes this young member of the Class of 1999; and time will tell which people and communities the power of that experience will touch, directly and indirectly, as she uses it to build her future. -

D Inosaur Paleobiology
Topics in Paleobiology The study of dinosaurs has been experiencing a remarkable renaissance over the past few decades. Scientifi c understanding of dinosaur anatomy, biology, and evolution has advanced to such a degree that paleontologists often know more about 100-million-year-old dinosaurs than many species of living organisms. This book provides a contemporary review of dinosaur science intended for students, researchers, and dinosaur enthusiasts. It reviews the latest knowledge on dinosaur anatomy and phylogeny, Brusatte how dinosaurs functioned as living animals, and the grand narrative of dinosaur evolution across the Mesozoic. A particular focus is on the fossil evidence and explicit methods that allow paleontologists to study dinosaurs in rigorous detail. Scientifi c knowledge of dinosaur biology and evolution is shifting fast, Dinosaur and this book aims to summarize current understanding of dinosaur science in a technical, but accessible, style, supplemented with vivid photographs and illustrations. Paleobiology Dinosaur The Topics in Paleobiology Series is published in collaboration with the Palaeontological Association, Paleobiology and is edited by Professor Mike Benton, University of Bristol. Stephen Brusatte is a vertebrate paleontologist and PhD student at Columbia University and the American Museum of Natural History. His research focuses on the anatomy, systematics, and evolution of fossil vertebrates, especially theropod dinosaurs. He is particularly interested in the origin of major groups such Stephen L. Brusatte as dinosaurs, birds, and mammals. Steve is the author of over 40 research papers and three books, and his work has been profi led in The New York Times, on BBC Television and NPR, and in many other press outlets. -

A Huge Predatory Dinosaur, Built to Swim
Science Now Discoveries from the world of science and medicine Spinosaurus: A huge predatory dinosaur, built to swim A digital skeletal reconstruction and transparent flesh outline of Spinosaurus aegyptiacus. The bones suggest this dinosaur was built to swim. Color codes show the origin of different parts of the digital skeletal model. (Model by Tyler Keillor, Lauren Conroy and Erin Fitzgerald, Ibrahim et al., Science/AAAS) By AMINA KHAN SEPTEMBER 11, 2014, 9:45 PM inosaurs ruled the land for millions of years. Now scientists have discovered a fearsome D species that could have wreaked havoc in its prehistoric waters. An unusual fossil whose parts were flung across two continents appears to be the first known semi-aquatic dinosaur, according to a report published Thursday by the journal Science. Measuring 9 feet longer than a Tyrannosaurus rex, the 95-million-year-old Spinosaurus aegyptiacus would have been the largest predatory dinosaur to walk the Earth. But it had several features that strongly suggest a life spent largely submerged in the water, including nostrils pushed toward the top of its skull and diagonally jutted teeth ideal for snapping up fish. “It was not a balancing, two-legged animal on land,” said study coauthor Paul Sereno, a paleontologist at the University of Chicago. “It would have been something very peculiar.” The differences between this Spinosaurus and other theropods are apparent from head to toe. Most theropods, like T. rex, ran on two powerful legs and had small, spindly arms. By contrast, the 50-foot-long Spinosaurus had muscular arms with blade-like claws that could have nabbed slippery prey, and shorter legs that were ill-equipped to walk on land. -

La Brea and Beyond: the Paleontology of Asphalt-Preserved Biotas
La Brea and Beyond: The Paleontology of Asphalt-Preserved Biotas Edited by John M. Harris Natural History Museum of Los Angeles County Science Series 42 September 15, 2015 Cover Illustration: Pit 91 in 1915 An asphaltic bone mass in Pit 91 was discovered and exposed by the Los Angeles County Museum of History, Science and Art in the summer of 1915. The Los Angeles County Museum of Natural History resumed excavation at this site in 1969. Retrieval of the “microfossils” from the asphaltic matrix has yielded a wealth of insect, mollusk, and plant remains, more than doubling the number of species recovered by earlier excavations. Today, the current excavation site is 900 square feet in extent, yielding fossils that range in age from about 15,000 to about 42,000 radiocarbon years. Natural History Museum of Los Angeles County Archives, RLB 347. LA BREA AND BEYOND: THE PALEONTOLOGY OF ASPHALT-PRESERVED BIOTAS Edited By John M. Harris NO. 42 SCIENCE SERIES NATURAL HISTORY MUSEUM OF LOS ANGELES COUNTY SCIENTIFIC PUBLICATIONS COMMITTEE Luis M. Chiappe, Vice President for Research and Collections John M. Harris, Committee Chairman Joel W. Martin Gregory Pauly Christine Thacker Xiaoming Wang K. Victoria Brown, Managing Editor Go Online to www.nhm.org/scholarlypublications for open access to volumes of Science Series and Contributions in Science. Natural History Museum of Los Angeles County Los Angeles, California 90007 ISSN 1-891276-27-1 Published on September 15, 2015 Printed at Allen Press, Inc., Lawrence, Kansas PREFACE Rancho La Brea was a Mexican land grant Basin during the Late Pleistocene—sagebrush located to the west of El Pueblo de Nuestra scrub dotted with groves of oak and juniper with Sen˜ora la Reina de los A´ ngeles del Rı´ode riparian woodland along the major stream courses Porciu´ncula, now better known as downtown and with chaparral vegetation on the surrounding Los Angeles. -

Dec 2001 Entire
P a l æ o n t o l o g i c a l S o c i e t y B u l l e t i n VOLUMEA 16 • NUMBER l 4 b e www.albertapaleo.org r t a DECEMBER 2001 ALBERTA PALÆONTOLOGICAL SOCIETY OFFICERS MEMBERSHIP: Any person with a sincere interest in President* Vaclav Marsovsky 547-0182 palaeontology is eligible to present their application for Vice-President* Dan Quinsey 247-3022 membership in the Society. (Please enclose membership Treasurer* (acting) Cindy Evans 285-0144 dues with your request for application.) Secretary* (acting)Dan Quinsey 247-3022 Single membership $15.00 annually Past-President* Wayne Braunberger 278-5154 Family or Institution $20.00 annually DIRECTORS Editor* Howard Allen 274-1858 THE BULLETIN WILL BE PUBLISHED Membership* Howard Allen 274-1858 QUARTERLY: March, June, September and Program Coordinator* Philip Benham 280-6283 December. Deadline for submitting material for Curator Ron Fortier 285-8041 publication is the 15th of the month prior to Librarian Mona Marsovsky 547-0182 publication. Events Coordinator* Keith Mychaluk 228-3211 Director at Large Dr. David Mundy 281-3668 Society Mailing Address: Social Director (position currently unfilled) Alberta Palaeontological Society APAC Representative† Vaclav Marsovsky 547-0182 P.O. Box 35111, Sarcee Postal Outlet * Officers and Directors marked with an asterisk are se- Calgary, Alberta, Canada T3E 7C7 nior board members for executive meeting quorum (Web: www.albertapaleo.org) purposes. † APAC is the Alberta Palaeontological Advisory Material for the Bulletin: Committee Howard Allen, Editor, APS 7828 Hunterslea Crescent, N.W. The Society was incorporated in 1986, as a non-profit Calgary, Alberta, Canada T2K 4M2 organization formed to: (E-mail: [email protected]) a. -

Notas Sobre Spinosauridae (Theropoda, Dinosauria) Notes on Spinosauridae (Theropoda, Dinosauria)
Anuário do Instituto de Geociências - UFRJ ISSN 0101-9759 Vol. 28-1 / 2005 p. 158-173 Notas Sobre Spinosauridae (Theropoda, Dinosauria) Notes on Spinosauridae (Theropoda, Dinosauria) Elaine Batista Machado1 & Alexander Wilhelm Armin Kellner2 1Setor de Paleovertebrados, Museu Nacional/UFRJ Quinta da Boa Vista s/n., São Cristóvão, Rio de Janeiro - RJ, CEP: 20940-040 e-mail: [email protected] 2Setor de Paleovertebrados, Museu Nacional/UFRJ Quinta da Boa Vista s/n., São Cristóvão, Rio de Janeiro - RJ, CEP: 20940-040 email: [email protected] Recebido: 08/11/2005 Aprovado: 19/11/2005 Resumo Os espinossaurídeos são encontrados na África, Europa, Ásia e Brasil em depósitos com idade variando do Jurássico Superior até o Cretáceo Superior, tendo sido descritos até a presente data oito espécies. Apesar desta grande distribuição temporal e geográfica, pouco se sabe sobre este grupo de dinossauros uma vez que a maior parte dos espécimes são incompletos. O clado Spinosauridae é dividido em Baryonychinae (Suchomimus + Baryonyx + Cristatosaurus) e Spinosaurinae (Spinosaurus+ Angaturama + Irritator). Apenas na África representantes destes dois clados foram encontrados. Uma possível explicação para a distribuição dos espinossaurídeos seria a dispersão de formas relacionadas a Baryonyx (o mais primitivo membro do grupo) da Europa para a África, onde surgiram os Spinosaurinae que posteriormente se distribuíram para a América do Sul. Palavras-chave: Dinosauria, Theropoda, Spinosauridae Abstract Spinosaur remains were collected in Africa, Europa, Asia and Brazil, coming from deposits with ages ranging from the Late Jurassic to the Late Cretaceous. Although having a wide distribution - both geographically and temporally - little is actually known about the group since most specimens are incomplete. -
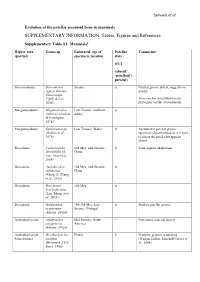
SUPPLEMENTARY INFORMATION: Tables, Figures and References
Samuels et al. Evolution of the patellar sesamoid bone in mammals SUPPLEMENTARY INFORMATION: Tables, Figures and References Supplementary Table S1: Mammals$ Higher taxa Genus sp. Estimated. age of Patellar Comments# (partial) specimen, location state 0/1/2 (absent/ ‘patelloid’/ present) Sinoconodonta Sinoconodon Jurassic 0 Patellar groove absent, suggests no rigneyi (Kielan- patella Jaworowska, Cifelli & Luo, Sinoconodon is included on our 2004) phylogeny within tritylodontids. Morganucodonta Megazostrodon Late Triassic, southern 0 rudnerae (Jenkins Africa & Parrington, 1976) Morganucodonta Eozostrodon sp. Late Triassic, Wales 0 Asymmetric patellar groove, (Jenkins et al., specimens disarticulated so it is hard 1976) to assess the patella but appears absent Docodonta Castorocauda 164 Mya, mid-Jurassic, 0 Semi-aquatic adaptations lutrasimilis (Ji, China Luo, Yuan et al., 2006) Docodonta Agilodocodon 164 Mya, mid-Jurassic, 0 scansorius China (Meng, Ji, Zhang et al., 2015) Docodonta Docofossor 160 Mya 0 brachydactylus (Luo, Meng, Ji et al., 2015) Docodonta Haldanodon 150-155 Mya, Late 0 Shallow patellar groove exspectatus Jurassic, Portugal (Martin, 2005b) Australosphenida Asfaltomylos Mid-Jurassic, South ? Postcranial material absent patagonicus America (Martin, 2005a) Australosphenida Ornithorhynchus Extant 2 Platypus, genome sequenced Monotremata anatinus (Warren, Hillier, Marshall Graves et (Herzmark, 1938; al., 2008) Rowe, 1988) Samuels et al. Australosphenida Tachyglossus + Extant 2 Echidnas Monotremata Zaglossus spp. (Herzmark, 1938; Rowe, 1988) Mammaliaformes Fruitafossor 150 Mya, Late Jurassic, 0 Phylogenetic status uncertain indet. windscheffeli (Luo Colorado & Wible, 2005) Mammaliaformes Volaticotherium Late Jurassic/Early ? Hindlimb material incomplete indet. antiquus (Meng, Cretaceous Hu, Wang et al., 2006) Eutriconodonta Jeholodens 120-125 Mya, Early 0 Poorly developed patellar groove jenkinsi (Ji, Luo Cretaceous, China & Ji, 1999) Eutriconodonta Gobiconodon spp. -

Triassic, Carnian) and the Early Evolution of Sauropodomorpha
A Basal Sauropodomorph (Dinosauria: Saurischia) from the Ischigualasto Formation (Triassic, Carnian) and the Early Evolution of Sauropodomorpha Ricardo N. Martinez*, Oscar A. Alcober Museo de Ciencias Naturales, San Juan, Argentina Abstract Background: The earliest dinosaurs are from the early Late Triassic (Carnian) of South America. By the Carnian the main clades Saurischia and Ornithischia were already established, and the presence of the most primitive known sauropodomorph Saturnalia suggests also that Saurischia had already diverged into Theropoda and Sauropodomorpha. Knowledge of Carnian sauropodomorphs has been restricted to this single species. Methodology/Principal Findings: We describe a new small sauropodomorph dinosaur from the Ischigualsto Formation (Carnian) in northwest Argentina, Panphagia protos gen. et sp. nov., on the basis of a partial skeleton. The genus and species are characterized by an anteroposteriorly elongated fossa on the base of the anteroventral process of the nasal; wide lateral flange on the quadrate with a large foramen; deep groove on the lateral surface of the lower jaw surrounded by prominent dorsal and ventral ridges; bifurcated posteroventral process of the dentary; long retroarticular process transversally wider than the articular area for the quadrate; oval scars on the lateral surface of the posterior border of the centra of cervical vertebrae; distinct prominences on the neural arc of the anterior cervical vertebra; distal end of the scapular blade nearly three times wider than the neck; scapular blade with an expanded posterodistal corner; and medial lamina of brevis fossa twice as wide as the iliac spine. Conclusions/Significance: We regard Panphagia as the most basal sauropodomorph, which shares the following apomorphies with Saturnalia and more derived sauropodomorphs: basally constricted crowns; lanceolate crowns; teeth of the anterior quarter of the dentary higher than the others; and short posterolateral flange of distal tibia. -

506 Kellner.Pmd
Arquivos do Museu Nacional, Rio de Janeiro, v.62, n.3, p.309-320, jul./set.2004 ISSN 0365-4508 ON A SEQUENCE OF SACROCAUDAL THEROPOD DINOSAUR VERTEBRAE FROM THE LOWER CRETACEOUS SANTANA FORMATION, NORTHEASTERN BRAZIL 1 (With 14 figures) JONATHAS DE SOUZA BITTENCOURT 2, 3 ALEXANDER WILHELM ARMIN KELLNER 2, 4 ABSTRACT: Besides being rare, most theropod remains from fossil deposits of Brazil are incomplete. Up to date the Romualdo Member (Aptian/Albian) of the Santana Formation yielded six theropod specimens. To those we add the description of a sequence of three posterior sacral and six anterior caudal vertebrae with three chevrons (MN 4743-V). Differences between MN 4743-V and members of the major theropod clades such as Ceratosauria, Allosauroidea and Coelorusauria do not allow its assignment to one of those groups. Instead, MN 4743-V is referred to the Spinosauroidea (Spinosauridae plus Torvosauridae), based on the presence of three robust laminae below the transverse process of the anterior caudals, which delimit three fossae. The lack of paired processes on the chevrons suggest that within Spinosauroidea MN 4743-V is a member of the Spinosauridae. This is congruent with previous findings of spinosaurids in the Romualdo Member. MN 4743-V differs from the remaining specimens that present sacrocaudal elements indicating the co-existence of at least five theropod species in this deposit. Key words: Spinosauroidea, Dinosauria, Santana Formation, Lower Cretaceous, Brazil. RESUMO: Sobre uma seqüência de vértebras sacrocaudais de um dinossauro terópode da Formação Santana, Cretáceo Inferior, Nordeste do Brasil. Restos de terópodes de depósitos fossilíferos brasileiros são raros, sendo a maioria dos espécimens incompletos. -

The Origin of the Dinosaurs El Origen De Los Dinosaurios
III Jornadas Internacionales sobre Paleontología de Dinosaurios y su Entorno Salas de los Infantes, Burgos The Origin of the Dinosaurs El origen de los dinosaurios M. J. Benton Department of Earth Sciences, University of Bristol, Bristol, BS8 1RJ, U.K. e-mail: [email protected] Recibido el 15 de diciembre de 2004, aceptado el 18 de diciembre de 2005. Abstract The origin of the dinosaurs has long been debated. There are two aspects, phylogenetic and ecological-evo- lutionary. Much of the phylogenetic confusion has been resolved by cladistic analysis of basal archosaurs which shows that the dinosaurs originated as part of a major clade Avemetatarsalia/ Ornithodira. Closest relatives of the dinosaurs are small Mid Triassic bipedal animals such as Marasuchus from Argentina. The basal avemetatarsalian is Scleromochlus from the Late Triassic of Scotland. The classic ecological-evolutionary model for the initial radiation of the dinosaurs had been that they competed gradually through the Triassic with precursor groups, and eventually prevailed. More detailed study of the timing of events suggests that the dinosaurs radiated opportunistically in a two-phase model, with expansion of herbivorous sauropodomorphs fi rst in the early Norian, and expansion of large theropods and ornithischians in the Early and Mid Jurassic. Both expansion phases followed extinction events. Key words: Dinosaur, Triassic, origin, opportunism, Eoraptor, Herrerasaurus. Resumen El origen de los dinosaurios ha sido debatido durante mucho tiempo. Hay dos puntos de vista, el fi logenético y el ecológico-evolutivo. La mayor parte de la confusión fi logenética ha sido resuelta por análisis cladístico en arco- saurios basales.