Abetalipoproteinemia [OMIM#200100]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Case with Dyskinesia Induced by Pramipexole, Pregabalin And
DO I:10.4274/Tnd.63497 Case Report / Olgu Sunumu A Case with Dyskinesia Induced by Pramipexole, Pregabalin and Gabapentin After Cardiopulmonary Resuscitation Kardiyopulmoner Resusitasyon Sonrası Pramipeksol, Pregabalin ve Gabapentin Kullanımının Tetiklediği Diskinezi Olgusu Dürdane Aksoy, Betül Çevik, Volkan Solmaz, Semiha Gülsüm Kurt, Orhan Sümbül Gaziosmanpaşa University School of Medicine, Department of Neurology, Tokat, Turkey Sum mary Drug-induced dyskinesias can be seen occasionally in clinical practice. Here we present a 70-year-old patient who developed a noticeable dyskinesia after the use of pramipexole, gabapentin, pregabalin respectively for his restless leg syndrome. Prior to this condition, he was hospitalized in intensive care unit for the myocardial infarction that required cardiopulmonary resuscitation, and he was discharged with no neurological deficits. The case presented here is a good example indicating the importance of being vigilant for drug-induced dyskinesias after hypoxic-ischemic encephalopathy, even though everything else seems right. (Turkish Journal of Neurology 2013; 19:148-150) Key Words: Hypoxic-ischemic encephalopathy, dyskinesia, pramipexole, gabapentin, pregabalin Özet İlaç kullanımı sonrasında ortaya çıkan hareket bozuklukları klinik pratikte zaman zaman karşılaştığımız sorunlardır. Burada kardiyopulmoner resüsitasyon gerektiren bir miyokard enfarktüsünün ardından bir süre yoğun bakımda kalan, nörolojik defisiti kalmadan iyileşen ancak daha sonra huzursuz bacak sendromu tedavisi için başlanan pramipeksol, -

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Iron Dysregulation in Movement Disorders
Neurobiology of Disease 46 (2012) 1–18 Contents lists available at SciVerse ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi Review Iron dysregulation in movement disorders Petr Dusek a,c, Joseph Jankovic a,⁎, Weidong Le b a Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, Houston, TX 77030, USA b Parkinson's Disease Research Laboratory, Department of Neurology, Baylor College of Medicine, Houston, TX 77030, USA c Department of Neurology and Center of Clinical Neuroscience, Charles University in Prague, 1st Faculty of Medicine and General University Hospital, Prague, Czech Republic article info abstract Article history: Iron is an essential element necessary for energy production, DNA and neurotransmitter synthesis, myelination Received 9 November 2011 and phospholipid metabolism. Neurodegeneration with brain iron accumulation (NBIA) involves several genetic Revised 22 December 2011 disorders, two of which, aceruloplasminemia and neuroferritinopathy, are caused by mutations in genes directly Accepted 31 December 2011 involved in iron metabolic pathway, and others, such as pantothenate-kinase 2, phospholipase-A2 and fatty acid Available online 12 January 2012 2-hydroxylase associated neurodegeneration, are caused by mutations in genes coding for proteins involved in phospholipid metabolism. Phospholipids are major constituents of myelin and iron accumulation has been linked Keywords: Iron to myelin derangements. Another group of NBIAs is caused by mutations in lysosomal enzymes or transporters Neurodegeneration such as ATP13A2, mucolipin-1 and possibly also β-galactosidase and α-fucosidase. Increased cellular iron uptake Dystonia in these diseases may be caused by impaired recycling of iron which normally involves lysosomes. -
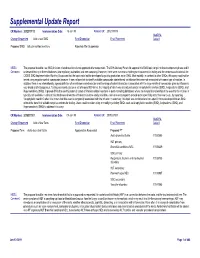
Detail Report
Supplemental Update Report CR Number: 2012319113 Implementation Date: 16-Jan-19 Related CR: 2012319113 MedDRA Change Requested Add a new SMQ Final Disposition Final Placement Code # Proposed SMQ Infusion related reactions Rejected After Suspension MSSO The proposal to add a new SMQ Infusion related reactions is not approved after suspension. The ICH Advisory Panel did approve this SMQ topic to go into the development phase and it Comment: underwent testing in three databases (two regulatory authorities and one company). However, there were numerous challenges encountered in testing and the consensus decision of the CIOMS SMQ Implementation Working Group was that the topic could not be developed to go into production as an SMQ. Most notably, in contrast to other SMQs, this query could not be tested using negative control compounds because it was not possible to identify suitable compounds administered via infusion that were not associated with some type of reaction. In addition, there is no internationally agreed definition of an infusion related reaction and the range of potential reactions associated with the large variety of compounds given by infusion is very broad and heterogenous. Testing was conducted on a set of around 500 terms, the majority of which was already included in Anaphylactic reaction (SMQ), Angioedema (SMQ), and Hypersensitivity (SMQ). It proved difficult to identify potential cases of infusion related reactions in post-marketing databases where the temporal relationship of the event to the infusion is typically not available. In clinical trial databases where this information is more easily available, users are encouraged to provide more specificity about the event, e.g., by reporting “Anaphylactic reaction” when it is known that this event is temporally associated with the infusion. -

Inherited Neuropathies
407 Inherited Neuropathies Vera Fridman, MD1 M. M. Reilly, MD, FRCP, FRCPI2 1 Department of Neurology, Neuromuscular Diagnostic Center, Address for correspondence Vera Fridman, MD, Neuromuscular Massachusetts General Hospital, Boston, Massachusetts Diagnostic Center, Massachusetts General Hospital, Boston, 2 MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology Massachusetts, 165 Cambridge St. Boston, MA 02114 and The National Hospital for Neurology and Neurosurgery, Queen (e-mail: [email protected]). Square, London, United Kingdom Semin Neurol 2015;35:407–423. Abstract Hereditary neuropathies (HNs) are among the most common inherited neurologic Keywords disorders and are diverse both clinically and genetically. Recent genetic advances have ► hereditary contributed to a rapid expansion of identifiable causes of HN and have broadened the neuropathy phenotypic spectrum associated with many of the causative mutations. The underlying ► Charcot-Marie-Tooth molecular pathways of disease have also been better delineated, leading to the promise disease for potential treatments. This chapter reviews the clinical and biological aspects of the ► hereditary sensory common causes of HN and addresses the challenges of approaching the diagnostic and motor workup of these conditions in a rapidly evolving genetic landscape. neuropathy ► hereditary sensory and autonomic neuropathy Hereditary neuropathies (HN) are among the most common Select forms of HN also involve cranial nerves and respiratory inherited neurologic diseases, with a prevalence of 1 in 2,500 function. Nevertheless, in the majority of patients with HN individuals.1,2 They encompass a clinically heterogeneous set there is no shortening of life expectancy. of disorders and vary greatly in severity, spanning a spectrum Historically, hereditary neuropathies have been classified from mildly symptomatic forms to those resulting in severe based on the primary site of nerve pathology (myelin vs. -

Neurological Disorders in Liver Transplant Candidates: Pathophysiology ☆ and Clinical Assessment
Transplantation Reviews 31 (2017) 193–206 Contents lists available at ScienceDirect Transplantation Reviews journal homepage: www.elsevier.com/locate/trre Neurological disorders in liver transplant candidates: Pathophysiology ☆ and clinical assessment Paolo Feltracco a,⁎, Annachiara Cagnin b, Cristiana Carollo a, Stefania Barbieri a, Carlo Ori a a Department of Medicine UO Anesthesia and Intensive Care, Padova University Hospital, Padova, Italy b Department of Neurosciences (DNS), University of Padova, Padova, Italy abstract Compromised liver function, as a consequence of acute liver insufficiency or severe chronic liver disease may be associated with various neurological syndromes, which involve both central and peripheral nervous system. Acute and severe hyperammoniemia inducing cellular metabolic alterations, prolonged state of “neuroinflamma- tion”, activation of brain microglia, accumulation of manganese and ammonia, and systemic inflammation are the main causative factors of brain damage in liver failure. The most widely recognized neurological complications of serious hepatocellular failure include hepatic encephalopathy, diffuse cerebral edema, Wilson disease, hepatic myelopathy, acquired hepatocerebral degeneration, cirrhosis-related Parkinsonism and osmotic demyelination syndrome. Neurological disorders affecting liver transplant candidates while in the waiting list may not only sig- nificantly influence preoperative morbidity and even mortality, but also represent important predictive factors for post-transplant neurological manifestations. -

Cerebellar Ataxia
CEREBELLAR ATAXIA Dr. Waqar Saeed Ziauddin Medical University, Karachi, Pakistan What is Ataxia? ■ Derived from a Greek word, ‘A’ : not, ‘Taxis’ : orderly Ataxia is defined as an inability to maintain normal posture and smoothness of movement. Types of Ataxia ■ Cerebellar Ataxia ■ Sensory Ataxia ■ Vestibular Ataxia Cerebellar Ataxia Cerebrocerebellum Spinocerebellum Vestibulocerebellum Vermis Planning and Equilibrium balance Posture, limb and initiating and posture eye movements movements Limb position, touch and pressure sensation Limb ataxia, Eye movement dysdiadochokinesia, disorders, Truncal and gait Dysmetria dysarthria nystagmus, VOR, ataxia hypotonia postural and gait. Gait ataxia Types of Cerebellar Ataxia • Vascular Acute Ataxia • Medications and toxins • Infectious etiologies • Atypical Infectious agents • Autoimmune disorders • Primary or metastatic tumors Subacute Ataxia • Paraneoplastic cerebellar degeneration • Alcohol abuse and Vitamin deficiencies • Systemic disorders • Autosomal Dominant Chronic • Autosomal recessive Progressive • X linked ataxias • Mitochondrial • Sporadic neurodegenerative diseases Vascular Ataxia ▪ Benedikt Syndrome It is a rare form of posterior circulation stroke of the brain. A lesion within the tegmentum of the midbrain can produce Benedikt Syndrome. Disease is characterized by ipsilateral third nerve palsy with contralateral hemitremor. Superior cerebellar peduncle and/or red nucleus damage in Benedikt Syndrome can further lead in to contralateral cerebellar hemiataxia. ▪ Wallenberg Syndrome In -

Studies on the Absorptive Defect for Triglyceride in Abetalipoproteinemia * P
Jownal of Clinical Investigation Vol. 46, No. 1, 1967 Studies on the Absorptive Defect for Triglyceride in Abetalipoproteinemia * P. 0. WAYS,t C. M. PARMENTIER, H. J. KAYDEN,4 J. W. JONES,§ D. R. SAUNDERS,|| AND C. E. RUBIN 1J (From the Department of Medicine, University of Washington School of Medicine, Seattle, Wash., and the Department of Medicine, New York University School of Medicine, New York, N. Y.) Summary. The nature of the gastrointestinal absorptive defect for triglycer- ide in three subjects with abetalipoproteinemia has been investigated by studying peroral biopsies of the gastrointestinal mucosa. The following con- clusions were reached. 1) In confirmation of other studies, the abnormal vacuoles within the du- odenal absorptive cells of these individuals were lipophilic. 2) On chemical analysis there was significantly more mucosal lipid than found in normal fasting specimens, and almost the entire increase was due to triglyceride. 3) This excess mucosal lipid was reduced by a low fat diet, but even after 34 days on such a diet there was still an excess of lipophilic material near the villus tip and increased quantities of total lipid and triglyceride when com- pared with material from normal subjects similarly treated. 4) Although there are demonstrable qualitative changes in mucosal and plasma lipids after an acute fat load, they are not quantitatively as great as in normal individuals. Fat balance studies and the qualitative changes in plasma and tissue lipids that do occur after more extended periods on different types of dietary fat do indicate that a considerable percentage of the dietary fat is assimilated. -

Bovine Milk Lipoprotein Lipase Transfers Tocopherol to Human Fibroblasts During Triglyceride Hydrolysis in Vitro Maret G
Bovine Milk Lipoprotein Lipase Transfers Tocopherol to Human Fibroblasts during Triglyceride Hydrolysis In Vitro Maret G. Traber, Thomas Olivecrona, and Herbert J. Kayden Department ofMedicine, New York University School ofMedicine, New York 10016; University of Umea, Umea, Sweden Abstract vitamin E, have minimal or absent neurological abnormalities, but those who have not been supplemented have a progressive Lipoprotein lipase appears to function as the mechanism by which dietary vitamin E (tocopherol) is transferred from chy- degeneration of the peripheral nervous system resulting in ataxia and characteristic pathologic changes in the central lomicrons to tissues. In patients with lipoprotein lipase defi- nervous system (5). These neuropathologic changes have been more than 85% of both the and ciency, circulating triglyceride observed in patients with cholestatic liver disease in whom the tocopherol is contained in the chylomicron fraction. The studies in low to absent levels of bile salts in the intestine results in an presented here show that the vitro addition of bovine milk inability to absorb vitamin E (3). Oral supplementation with to in the of lipoprotein lipase (lipase) chylomicrons presence pharmacologic amounts of the vitamin (6), or or fibroblasts bovine albumin pagenteral human erythrocytes (and serum administration of the vitamin (when there is a complete resulted in the hydrolysis of triglyceride and the [BSAJ) the absence of intestinal bile) (7) results in the prevention of transfer of both acids and to the in the fatty tocopherol cells; further of the nervous system. The studies in absence of no in cellular was deterioration lipase, increase tocopherol these two groups ofpatients have demonstrated the importance The incubation was to include detectable. -

GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies
International Journal of Molecular Sciences Review GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies Andrés Felipe Leal 1 , Eliana Benincore-Flórez 1, Daniela Solano-Galarza 1, Rafael Guillermo Garzón Jaramillo 1 , Olga Yaneth Echeverri-Peña 1, Diego A. Suarez 1,2, Carlos Javier Alméciga-Díaz 1,* and Angela Johana Espejo-Mojica 1,* 1 Institute for the Study of Inborn Errors of Metabolism, Faculty of Science, Pontificia Universidad Javeriana, Bogotá 110231, Colombia; [email protected] (A.F.L.); [email protected] (E.B.-F.); [email protected] (D.S.-G.); [email protected] (R.G.G.J.); [email protected] (O.Y.E.-P.); [email protected] (D.A.S.) 2 Faculty of Medicine, Universidad Nacional de Colombia, Bogotá 110231, Colombia * Correspondence: [email protected] (C.J.A.-D.); [email protected] (A.J.E.-M.); Tel.: +57-1-3208320 (ext. 4140) (C.J.A.-D.); +57-1-3208320 (ext. 4099) (A.J.E.-M.) Received: 6 July 2020; Accepted: 7 August 2020; Published: 27 August 2020 Abstract: GM2 gangliosidoses are a group of pathologies characterized by GM2 ganglioside accumulation into the lysosome due to mutations on the genes encoding for the β-hexosaminidases subunits or the GM2 activator protein. Three GM2 gangliosidoses have been described: Tay–Sachs disease, Sandhoff disease, and the AB variant. Central nervous system dysfunction is the main characteristic of GM2 gangliosidoses patients that include neurodevelopment alterations, neuroinflammation, and neuronal apoptosis. Currently, there is not approved therapy for GM2 gangliosidoses, but different therapeutic strategies have been studied including hematopoietic stem cell transplantation, enzyme replacement therapy, substrate reduction therapy, pharmacological chaperones, and gene therapy. -

Anatomical, Clinical, and Electrodiagnostic Features of Radial Neuropathies
Anatomical, Clinical, and Electrodiagnostic Features of Radial Neuropathies a, b Leo H. Wang, MD, PhD *, Michael D. Weiss, MD KEYWORDS Radial Posterior interosseous Neuropathy Electrodiagnostic study KEY POINTS The radial nerve subserves the extensor compartment of the arm. Radial nerve lesions are common because of the length and winding course of the nerve. The radial nerve is in direct contact with bone at the midpoint and distal third of the humerus, and therefore most vulnerable to compression or contusion from fractures. Electrodiagnostic studies are useful to localize and characterize the injury as axonal or demyelinating. Radial neuropathies at the midhumeral shaft tend to have good prognosis. INTRODUCTION The radial nerve is the principal nerve in the upper extremity that subserves the extensor compartments of the arm. It has a long and winding course rendering it vulnerable to injury. Radial neuropathies are commonly a consequence of acute trau- matic injury and only rarely caused by entrapment in the absence of such an injury. This article reviews the anatomy of the radial nerve, common sites of injury and their presentation, and the electrodiagnostic approach to localizing the lesion. ANATOMY OF THE RADIAL NERVE Course of the Radial Nerve The radial nerve subserves the extensors of the arms and fingers and the sensory nerves of the extensor surface of the arm.1–3 Because it serves the sensory and motor Disclosures: Dr Wang has no relevant disclosures. Dr Weiss is a consultant for CSL-Behring and a speaker for Grifols Inc. and Walgreens. He has research support from the Northeast ALS Consortium and ALS Therapy Alliance. -

COVID-19 Mrna Pfizer- Biontech Vaccine Analysis Print
COVID-19 mRNA Pfizer- BioNTech Vaccine Analysis Print All UK spontaneous reports received between 9/12/20 and 22/09/21 for mRNA Pfizer/BioNTech vaccine. A report of a suspected ADR to the Yellow Card scheme does not necessarily mean that it was caused by the vaccine, only that the reporter has a suspicion it may have. Underlying or previously undiagnosed illness unrelated to vaccination can also be factors in such reports. The relative number and nature of reports should therefore not be used to compare the safety of the different vaccines. All reports are kept under continual review in order to identify possible new risks. Report Run Date: 24-Sep-2021, Page 1 Case Series Drug Analysis Print Name: COVID-19 mRNA Pfizer- BioNTech vaccine analysis print Report Run Date: 24-Sep-2021 Data Lock Date: 22-Sep-2021 18:30:09 MedDRA Version: MedDRA 24.0 Reaction Name Total Fatal Blood disorders Anaemia deficiencies Anaemia folate deficiency 1 0 Anaemia vitamin B12 deficiency 2 0 Deficiency anaemia 1 0 Iron deficiency anaemia 6 0 Anaemias NEC Anaemia 97 0 Anaemia macrocytic 1 0 Anaemia megaloblastic 1 0 Autoimmune anaemia 2 0 Blood loss anaemia 1 0 Microcytic anaemia 1 0 Anaemias haemolytic NEC Coombs negative haemolytic anaemia 1 0 Haemolytic anaemia 6 0 Anaemias haemolytic immune Autoimmune haemolytic anaemia 9 0 Anaemias haemolytic mechanical factor Microangiopathic haemolytic anaemia 1 0 Bleeding tendencies Haemorrhagic diathesis 1 0 Increased tendency to bruise 35 0 Spontaneous haematoma 2 0 Coagulation factor deficiencies Acquired haemophilia