Identification of Genomic Aberrations by Array Comparative Genomic Hybridization in Patients with Aortic Dissections
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Gene Expression Is Related to Parental Origin and Regional Coordinate Control
Journal of Human Genetics (2009) 54, 193–198 & 2009 The Japan Society of Human Genetics All rights reserved 1434-5161/09 $32.00 www.nature.com/jhg ORIGINAL ARTICLE William’s syndrome: gene expression is related to parental origin and regional coordinate control Jeremy C Collette1, Xiao-Ning Chen1, Debra L Mills2, Albert M Galaburda3, Allan L Reiss4, Ursula Bellugi5 and Julie R Korenberg1,6 William’s syndrome (WS) features a spectrum of neurocognitive and behavioral abnormalities due to a rare 1.5 MB deletion that includes about 24–28 genes on chromosome band 7q11.23. Study of the expression of these genes from the single normal copy provides an opportunity to elucidate the genetic and epigenetic controls on these genes as well as their roles in both WS and normal brain development and function. We used quantitative RT-PCR to determine the transcriptional level of 14 WS gene markers in a cohort of 77 persons with WS and 48 normal controls. Results reported here: (1) show that the expression of the genes deleted in WS is decreased in some but not all cases, (2) demonstrate that the parental origin of the deletion contributes to the level of expression of GTF2I independently of age and gender and (3) indicate that the correlation of expression between GTF2I and some other genes in the WS region differs in WS subjects and normal controls, which in turn points toward a regulatory role for this gene. Interspecies comparisons suggest GTF2I may play a key role in normal brain development. Journal of Human Genetics (2009) 54, 193–198; doi:10.1038/jhg.2009.5; published online 13 March 2009 Keywords: William’s syndrome; gene expression; RT-PCR; parental origin; GTF2I INTRODUCTION As an approach toward understanding the role of the deleted genes William’s syndrome (WS) is a neurogenetic disorder affecting human in WS, we have characterized WS subjects according to genetic, social/ development and adult cognition. -
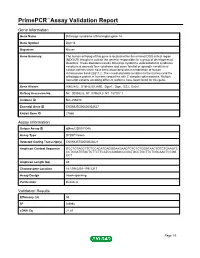
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DiGeorge syndrome critical region gene 14 Gene Symbol Dgcr14 Organism Mouse Gene Summary The human ortholog of this gene is located within the minimal DGS critical region (MDGCR) thought to contain the gene(s) responsible for a group of developmental disorders. These disorders include DiGeorge syndrome velocardiofacial syndrome conotruncal anomaly face syndrome and some familial or sporadic conotruncal cardiac defects which have been associated with microdeletion of human chromosome band 22q11.2. The encoded protein localizes to the nucleus and the orthologous protein in humans co-purifies with C complex spliceosomes. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases AI462402, D16H22S1269E, Dgcr1, Dgsi, ES2, Es2el RefSeq Accession No. NC_000082.6, NT_039624.8, NT_187007.1 UniGene ID Mm.256480 Ensembl Gene ID ENSMUSG00000003527 Entrez Gene ID 27886 Assay Information Unique Assay ID qMmuCID0011048 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000003621 Amplicon Context Sequence GCCTGTAGCTTCTCCACATCAGGGAAGAAGTCTCTCTGGATAACTGTCTGAAGTC CCTCGATGTACTCTTCTTCATCCAGGACCCGCTGCCTGCTTCTCGCAACTCCGG CCT Amplicon Length (bp) 82 Chromosome Location 16:17910201-17911217 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9994 cDNA Cq 21.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79.5 gDNA Cq 23.94 Specificity (%) 100 Information to assist with data interpretation is provided -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Supplemental Information Proximity Interactions Among Centrosome
Current Biology, Volume 24 Supplemental Information Proximity Interactions among Centrosome Components Identify Regulators of Centriole Duplication Elif Nur Firat-Karalar, Navin Rauniyar, John R. Yates III, and Tim Stearns Figure S1 A Myc Streptavidin -tubulin Merge Myc Streptavidin -tubulin Merge BirA*-PLK4 BirA*-CEP63 BirA*- CEP192 BirA*- CEP152 - BirA*-CCDC67 BirA* CEP152 CPAP BirA*- B C Streptavidin PCM1 Merge Myc-BirA* -CEP63 PCM1 -tubulin Merge BirA*- CEP63 DMSO - BirA* CEP63 nocodazole BirA*- CCDC67 Figure S2 A GFP – + – + GFP-CEP152 + – + – Myc-CDK5RAP2 + + + + (225 kDa) Myc-CDK5RAP2 (216 kDa) GFP-CEP152 (27 kDa) GFP Input (5%) IP: GFP B GFP-CEP152 truncation proteins Inputs (5%) IP: GFP kDa 1-7481-10441-1290218-1654749-16541045-16541-7481-10441-1290218-1654749-16541045-1654 250- Myc-CDK5RAP2 150- 150- 100- 75- GFP-CEP152 Figure S3 A B CEP63 – – + – – + GFP CCDC14 KIAA0753 Centrosome + – – + – – GFP-CCDC14 CEP152 binding binding binding targeting – + – – + – GFP-KIAA0753 GFP-KIAA0753 (140 kDa) 1-496 N M C 150- 100- GFP-CCDC14 (115 kDa) 1-424 N M – 136-496 M C – 50- CEP63 (63 kDa) 1-135 N – 37- GFP (27 kDa) 136-424 M – kDa 425-496 C – – Inputs (2%) IP: GFP C GFP-CEP63 truncation proteins D GFP-CEP63 truncation proteins Inputs (5%) IP: GFP Inputs (5%) IP: GFP kDa kDa 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl Myc- 150- Myc- 100- CCDC14 KIAA0753 100- 100- 75- 75- GFP- GFP- 50- CEP63 50- CEP63 37- 37- Figure S4 A siCtl -

The P53/P73 - P21cip1 Tumor Suppressor Axis Guards Against Chromosomal Instability by Restraining CDK1 in Human Cancer Cells
Oncogene (2021) 40:436–451 https://doi.org/10.1038/s41388-020-01524-4 ARTICLE The p53/p73 - p21CIP1 tumor suppressor axis guards against chromosomal instability by restraining CDK1 in human cancer cells 1 1 2 1 2 Ann-Kathrin Schmidt ● Karoline Pudelko ● Jan-Eric Boekenkamp ● Katharina Berger ● Maik Kschischo ● Holger Bastians 1 Received: 2 July 2020 / Revised: 2 October 2020 / Accepted: 13 October 2020 / Published online: 9 November 2020 © The Author(s) 2020. This article is published with open access Abstract Whole chromosome instability (W-CIN) is a hallmark of human cancer and contributes to the evolvement of aneuploidy. W-CIN can be induced by abnormally increased microtubule plus end assembly rates during mitosis leading to the generation of lagging chromosomes during anaphase as a major form of mitotic errors in human cancer cells. Here, we show that loss of the tumor suppressor genes TP53 and TP73 can trigger increased mitotic microtubule assembly rates, lagging chromosomes, and W-CIN. CDKN1A, encoding for the CDK inhibitor p21CIP1, represents a critical target gene of p53/p73. Loss of p21CIP1 unleashes CDK1 activity which causes W-CIN in otherwise chromosomally stable cancer cells. fi Vice versa 1234567890();,: 1234567890();,: Consequently, induction of CDK1 is suf cient to induce abnormal microtubule assembly rates and W-CIN. , partial inhibition of CDK1 activity in chromosomally unstable cancer cells corrects abnormal microtubule behavior and suppresses W-CIN. Thus, our study shows that the p53/p73 - p21CIP1 tumor suppressor axis, whose loss is associated with W-CIN in human cancer, safeguards against chromosome missegregation and aneuploidy by preventing abnormally increased CDK1 activity. -

A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L
www.nature.com/scientificreports OPEN A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L. Patel-Murray1, Miriam Adam2, Nhan Huynh2, Brook T. Wassie2, Pamela Milani2 & Ernest Fraenkel 2,3* High-throughput screening and gene signature analyses frequently identify lead therapeutic compounds with unknown modes of action (MoAs), and the resulting uncertainties can lead to the failure of clinical trials. We developed an approach for uncovering MoAs through an interpretable machine learning model of transcriptomics, epigenomics, metabolomics, and proteomics. Examining compounds with benefcial efects in models of Huntington’s Disease, we found common MoAs for compounds with unrelated structures, connectivity scores, and binding targets. The approach also predicted highly divergent MoAs for two FDA-approved antihistamines. We experimentally validated these efects, demonstrating that one antihistamine activates autophagy, while the other targets bioenergetics. The use of multiple omics was essential, as some MoAs were virtually undetectable in specifc assays. Our approach does not require reference compounds or large databases of experimental data in related systems and thus can be applied to the study of agents with uncharacterized MoAs and to rare or understudied diseases. Unknown modes of action of drug candidates can lead to unpredicted consequences on efectiveness and safety. Computational methods, such as the analysis of gene signatures, and high-throughput experimental methods have accelerated the discovery of lead compounds that afect a specifc target or phenotype1–3. However, these advances have not dramatically changed the rate of drug approvals. Between 2000 and 2015, 86% of drug can- didates failed to earn FDA approval, with toxicity or a lack of efcacy being common reasons for their clinical trial termination4,5. -

Cep57, a NEDD1-Binding Pericentriolar Material Component, Is Essential for Spindle Pole Integrity
Cell Research (2012) :1-12. © 2012 IBCB, SIBS, CAS All rights reserved 1001-0602/12 $ 32.00 npg ORIGINAL ARTICLE www.nature.com/cr Cep57, a NEDD1-binding pericentriolar material component, is essential for spindle pole integrity Qixi Wu1, *, Runsheng He1, *, Haining Zhou1, Albert CH Yu2, Bo Zhang1, Junlin Teng1, Jianguo Chen1, 3 1The State Key Laboratory of Biomembrane and Membrane Bioengineering and The Key Laboratory of Cell Proliferation and Dif- ferentiation of Ministry of Education, College of Life Sciences, Peking University, Beijing 100871, China; 2Department of Neurobi- ology, Neuroscience Research Institute, School of Basic Medical Sciences, Peking University, Beijing 100191, China; 3The Center for Theoretical Biology, Peking University, Beijing 100871, China Formation of a bipolar spindle is indispensable for faithful chromosome segregation and cell division. Spindle in- tegrity is largely dependent on the centrosome and the microtubule network. Centrosome protein Cep57 can bundle microtubules in mammalian cells. Its related protein (Cep57R) in Xenopus was characterized as a stabilization factor for microtubule-kinetochore attachment. Here we show that Cep57 is a pericentriolar material (PCM) component. Its interaction with NEDD1 is necessary for the centrosome localization of Cep57. Depletion of Cep57 leads to unaligned chromosomes and a multipolar spindle, which is induced by PCM fragmentation. In the absence of Cep57, cen- trosome microtubule array assembly activity is weakened, and the spindle length and microtubule density decrease. As a spindle microtubule-binding protein, Cep57 is also responsible for the proper organization of the spindle micro- tubule and localization of spindle pole focusing proteins. Collectively, these results suggest that Cep57, as a NEDD1- binding centrosome component, could function as a spindle pole- and microtubule-stabilizing factor for establishing robust spindle architecture. -

CRNKL1 Is a Highly Selective Regulator of Intron-Retaining HIV-1 and Cellular Mrnas
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 CRNKL1 is a highly selective regulator of intron-retaining HIV-1 and cellular mRNAs 2 3 4 Han Xiao1, Emanuel Wyler2#, Miha Milek2#, Bastian Grewe3, Philipp Kirchner4, Arif Ekici4, Ana Beatriz 5 Oliveira Villela Silva1, Doris Jungnickl1, Markus Landthaler2,5, Armin Ensser1, and Klaus Überla1* 6 7 1 Institute of Clinical and Molecular Virology, University Hospital Erlangen, Friedrich-Alexander 8 Universität Erlangen-Nürnberg, Erlangen, Germany 9 2 Berlin Institute for Medical Systems Biology, Max-Delbrück-Center for Molecular Medicine in the 10 Helmholtz Association, Robert-Rössle-Strasse 10, 13125, Berlin, Germany 11 3 Department of Molecular and Medical Virology, Ruhr-University, Bochum, Germany 12 4 Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander Universität Erlangen- 13 Nürnberg, Erlangen, Germany 14 5 IRI Life Sciences, Institute für Biologie, Humboldt Universität zu Berlin, Philippstraße 13, 10115, Berlin, 15 Germany 16 # these two authors contributed equally 17 18 19 *Corresponding author: 20 Prof. Dr. Klaus Überla 21 Institute of Clinical and Molecular Virology, University Hospital Erlangen 22 Friedrich-Alexander Universität Erlangen-Nürnberg 23 Schlossgarten 4, 91054 Erlangen 24 Germany 25 Tel: (+49) 9131-8523563 26 e-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Salinity Regulates Claudin Mrna and Protein Expression in the Teleost Gill
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Am J Physiol Regul Integr Comp Physiol 294: R1004–R1014, 2008. provided by University of Southern Denmark Research Output First published January 9, 2008; doi:10.1152/ajpregu.00112.2007. Salinity regulates claudin mRNA and protein expression in the teleost gill Christian K. Tipsmark,* David A. Baltzegar,* Ozkan Ozden, Brenda J. Grubb, and Russell J. Borski Department of Zoology, North Carolina State University, Raleigh, North Carolina Submitted 14 February 2007; accepted in final form 19 December 2007 Tipsmark CK, Baltzegar DA, Ozden O, Grubb BJ, Borski RJ. The biochemical basis for changes in water permeability may Salinity regulates claudin mRNA and protein expression in the teleost entail the robust downregulation of gill aquaporin mRNA and gill. Am J Physiol Regul Integr Comp Physiol 294: R1004–R1014, 2008. protein expression that accompanies SW acclimation in eel (6, First published January 9, 2008; doi:10.1152/ajpregu.00112.2007.—The 21). Although osmotic permeability decreases in SW, the teleost gill carries out NaCl uptake in freshwater (FW) and NaCl conductance and short-circuit current is found to be larger in a excretion in seawater (SW). This transformation with salinity requires close regulation of ion transporter capacity and epithelial permeabil- SW teleost compared with a FW fish (8, 27). A simultaneous change in the ultrastructure of tight junctions (TJs) is also seen Downloaded from ity. This study investigates the regulation of tight-junctional claudins ϩ during salinity acclimation in fish. We identified claudin 3- and (28). Secretion of Na is thought to involve a paracellular claudin 4-like immunoreactive proteins and examined their expression path confined to thin “leaky” TJs that occur in SW between and that of select ion transporters by performing Western blot in mature chloride cells and accessory cells (19, 36), and this tilapia (Oreochromis mossambicus) gill during FW and SW acclima- may contribute to the relatively high ionic permeability of tion.