Mechanisms of Chemical Generation of Volatile Hydrides for Trace Element Determination (IUPAC Technical Report)*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
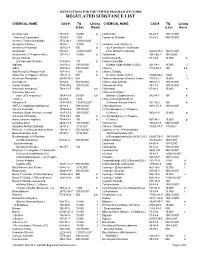
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties
THE JOURNAL OF ALTERNATIVE AND COMPLEMENTARY MEDICINE Volume 10, Number 2, 2004, pp. 337–344 ©Mary Ann Liebert, Inc. Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties BONNIEJ. KAPLAN, Ph.D., 1 W. WESLEYPARISH, Ph.D., 2 G. MERRILLANDRUS, Ph.D., 2 J. STEVENA. SIMPSON, Ph.D., M.D., 3 and CATHERINEJ. FIELD, Ph.D., R.D. 4 ABSTRACT This paper reviews the history, chemistry, safety, toxicity, and anticancer effects of the organogermanium compound bis (2-carboxyethylgermanium) sesquioxide (CEGS). A companion review follows, discussing the inaccuracies in the scientific record that have prematurely terminated research on clinical uses of CEGS. CEGS is a unique organogermanium compound first made by Mironov and coworkers in Russia and, shortly there- after, popularized by Asai and his colleagues in Japan. Low concentrations of germanium occur in nearly all soils, plants and animal life; natural occurrence of the CEGS form is postulated but not yet demonstrated. The literature demonstrating its anticancer effect is particularly strong: CEGS induces interferon- g (IFN-g), en- hances natural killer cell activity, and inhibits tumor and metastatic growth—effects often detectable after a single oral dose. In addition, oral consumption of CEGS is readily assimilated and rapidly cleared from the body without evidence of toxicity. Given these findings, the absence of human clinical trials of CEGS is un- expected. Possible explanations of why the convincing findings from animal research have not been used to support clinical trials are discussed. Clinical trials on CEGS are recommended. INTRODUCTION bispropionic acid; 3-oxygermylpropionic acid polymer; poly- trans-(2-carboxyethyl) germasesquioxane); proxigerma- n general, dietary supplements are an underinvestigated nium; repagermanium; and Serocion. -

Risto Laitinen/August 4, 2016 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structur
Approved Minutes, Busan 2015 Risto Laitinen/August 4, 2016 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Approved Minutes of Division Committee Meeting in Busan, Korea, 8–9 August, 2015 1. Welcome, introductory remarks and housekeeping announcements Karl-Heinz Hellwich (KHH) welcomed everybody to the meeting, extending a special welcome to those who were attending the Division Committee meeting for the first time. He described house rules and arrangements during the meeting. KHH also regretfully reported that it has come to his attention that since the Bangor meeting in August 2014, Prof. Derek Horton (Member, Division VIII task groups on Carbohydrate and Flavonoids nomenclature; Associate Member, IUBMB-IUPAC Joint Commission on Biochemical Nomenclature) and Dr. Libuse Goebels, Member of the former Commission on Nomenclature of Organic Chemistry) have passed away. The meeting attendees paid a tribute to their memory by a moment of silence. 2. Attendance and apologies Present: Karl-Heinz Hellwich (president, KHH) , Risto Laitinen (acting secretary, RSL), Richard Hartshorn (past-president, RMH), Michael Beckett (MAB), Alan Hutton (ATH), Gerry P. Moss (GPM), Michelle Rogers (MMR), Jiří Vohlídal (JV), Andrey Yerin (AY) Observers: Leah McEwen (part time, chair of proposed project, LME), Elisabeth Mansfield (task group chair, EM), Johan Scheers (young observer, day 1; JS), Prof. Kazuyuki Tatsumi (past- president of the union, part of day 2) Apologies: Ture Damhus (secretary, TD), Vefa Ahsen, Kirill Degtyarenko, Gernot Eller, Mohammed Abul Hashem, Phil Hodge (PH), Todd Lowary, József Nagy, Ebbe Nordlander (EN), Amélia Pilar Rauter (APR), Hinnerk Rey (HR), John Todd, Lidija Varga-Defterdarović. -

Boranes: Physical & Chemical Properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-In
Boranes: Physical & chemical properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-in-Chief. International Labor Organization, Geneva. 2011. Chemical Name Colour/Form Boiling Point Melting Molecular Solubility in Relative Density Relative Vapour Inflam. Flash Auto CAS-Number (°C) Point (°C) Weight Water (water=1) Vapour Pressure/ Limits Point (°C) Ignition Density (Kpa) Point (°C) (air=1) BORON polymorphic: alpha- 2550 2300 10.81 insol Amorphous, 1.56x 580 3 -5 7440-42-8 rhombohedral form, clear 2.3 g/cm ; 10 red crystals; beta- alpha-- @ 2140 °C rhombohedral form, black; rhombohedral, - alpha-tetragonal form, 2.46 g/cm3; - black, opaque crystals with alpha-- metallic luster; amorphous tetragonal, - form, black or dark brown 2.31 g/cm3; - powder; other crystal beta-rhom- forms known bohedral, - 2.35 g/cm3 BORIC ACID, DISODIUM powder or glass-like 1575 741 201.3 2.56 g/100 g 2.367 SALT plates; white, free-flowing 1330-43-4 crystals; light grey solid BORON OXIDE rhombic crystals; 1860 450 69.6 2.77 g/100 g 1.8 1303-86-2 colourless, (amorphous); semitransparent lumps or 2.46 hard, white crystals (crystalline) BORON TRIBROMIDE colourless liquid 90 -46.0 250.57 reacts 2.6431 8.6 5.3 10294-33-4 @ 18.4 °C/4 °C @ 14 °C BORON TRICHLORIDE 12.5 -107 117.16 1.35 4.03 2.99 Pa 10294-34-5 @ 12 °C/4 @ 12.4 °C BORON TRIFLUORIDE colourless gas -99.9 -126.8 67.82 reacts 3.08g/1.57 l 2.4 10 mm Hg 7637-07-2 @ 4 °C @ -141 °C. -

Germane 99.99+%
GEG5001 - GERMANE 99.99+% GERMANE 99.99+% Safety Data Sheet GEG5001 Date of issue: 01/05/2015 Version: 1.0 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Product form : Substance Physical state : Gas Substance name : GERMANE 99.99+% Product code : GEG5001 Formula : GeH4 Synonyms : MONOGERMANE; GERMANIUM HYDRIDE; GERMANIUM TETRAHYDRIDE Chemical family : GERMANE 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Chemical intermediate For research and industrial use only 1.3. Details of the supplier of the safety data sheet GELEST, INC. 11 East Steel Road Morrisville, PA 19067 USA T 215-547-1015 - F 215-547-2484 - (M-F): 8:00 AM - 5:30 PM EST [email protected] - www.gelest.com 1.4. Emergency telephone number Emergency number : CHEMTREC: 1-800-424-9300 (USA); +1 703-527-3887 (International) SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification (GHS-US) Flam. Gas 1 H220 Liquefied gas H280 Acute Tox. 2 (Inhalation:gas) H330 Eye Irrit. 2A H319 STOT SE 3 H335 Full text of H-phrases: see section 16 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS04 GHS06 GHS07 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H220 - Extremely flammable gas H280 - Contains gas under pressure; may explode if heated H319 - Causes serious eye irritation H330 - Fatal if inhaled H335 - May cause respiratory irritation Precautionary statements (GHS-US) : P284 - [In case of inadequate ventilation] wear respiratory protection P280 - Wear protective gloves/protective clothing/eye protection/face protection P260 - Do not breathe gas P264 - Wash hands thoroughly after handling P310 - Immediately call a doctor P210 - Keep away from heat, open flames, sparks. -

Solid-State Structures of the Covalent Hydrides Germane and Stannane
Edinburgh Research Explorer Solid-state structures of the covalent hydrides germane and stannane Citation for published version: Maley, IJ, Brown, DH, Ibberson, RM & Pulham, CR 2008, 'Solid-state structures of the covalent hydrides germane and stannane', Acta Crystallographica Section B - Structural Science, vol. 64, no. Pt 3, pp. 312-7. https://doi.org/10.1107/S0108768108010379 Digital Object Identifier (DOI): 10.1107/S0108768108010379 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Acta Crystallographica Section B - Structural Science Publisher Rights Statement: Copyright © 2008 International Union of Crystallography; all rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 electronic reprint Acta Crystallographica Section B Structural Science, Crystal Engineering and Materials ISSN 2052-5192 Solid-state structures of the covalent hydrides germane and stannane Iain J. Maley, Daniel H. Brown, Richard M. Ibberson and Colin R. Pulham Acta Cryst. (2008). B64, 312–317 Copyright c International Union of Crystallography Author(s) of this paper may load this reprint on their own web site or institutional repository provided that this cover page is retained. -

Hazardous Material Inventory Statement
City of Brooklyn Park FIRE DEPARTMENT 5200 - 85th Avenue North Brooklyn Park MN 55443 Phone: (763)493-8020 Fax: (763) 493-8391 Hazardous Materials Inventory Statement Users Guide A separate inventory statement shall be provided for each building. An amended inventory statement shall be provided within 30 days of the storage of any hazardous materials or plastics that changes or adds a hazard class or which is sufficient in quantity to cause an increase in the quantity which exceeds 5 percent for any hazard class. The hazardous materials inventory statement shall list by hazard class categories. Each grouping shall provide the following information for each hazardous material listed for that group including a total quantity for each group of hazard class. 1. Hazard class. (See attached Hazardous Materials Categories Listing) 2. Common or trade name. 3. Chemical Abstract Service Number (CAS number) found in 29 Code of Federal Regulations (C.F.R.). 4. Whether the material is pure or a mixture, and whether the material is a solid, liquid or gas 5. Maximum aggregate quantity stored at any one time. 6. Maximum aggregate quantity In-Use (Open to atmosphere) at any one time. 7. Maximum aggregate quantity In-Use (Closed to atmosphere) at any one time. 8. Storage conditions related to the storage type, high-pile, encapsulated, non-encapsulated. Attached is a listing of categories that all materials need to be organized to. Definitions of these categories are also attached for your use. At the end of this packet are blank forms for completing this project. For questions regarding Hazardous Materials Inventory Statement contact the Fire Department at 763-493-8020. -

Search for New Three-, Four-Component Petasis, Passerini
J. Chem. Chem. Eng. 8 (2014) 428-432 D DAVID PUBLISHING Search for New Three-, Four-Component Petasis, Passerini, Hantzsch, Kabachnic-Fields, Ugi Reactions with Organic Compounds of Phosphorus, Arsenic, Antimony and Bismuth Aibassov Erkin Zhakenovich*, Yemelyanova Valentina Stepanovna, Shakieva Tatyana Vladimirovna, Tussupbaev Nessipbay Kuandykovich and Blagikh Evgeniy Vladimirovich Research Institute of New Chemical Technologies and Materials, Kazakh National University Al-Farabi, Almaty 005012, Kazakhstan Received: November 29, 2013 / Accepted: December 18, 2013 / Published: April 25, 2014. Abstract: It is discovered a new three-, four-component Petasis, Passerini, Hantzsch, Kabachnic-Fields, Ugi reactions with of arsine, stibine and bismuthine in organometallic chemistry. Modifications were replaced to a nitrogen atom of classical reactions of atoms of phosphorus, arsenic, antimony and bismuth. It has been proposed a new mechanism for possible reactions. Key words: Petasis, Passerini, Hantzsch, Kabachnic-Fields, Ugi reactions, phosphorus, arsenic, antimony, bismuth. 1. Introduction four-component reactions Petasis, Passerini, Hantzsch, Kabachnic-Fields, Ugi reactions with organic In recent years, it has been actively investigated compounds of phosphorus, arsenic, antimony and nucleophilic reaction organilgalogenidami phosphine bismuth. and with electrophilic geterilalkenami allowing to obtain primary, secondary and tertiary phosphines, 2. Theory selectively and with high yield. Most of these It is known that the Petasis reaction is a MCR reactions are realized under mild conditions at a (multi component reaction) that enables the pressure atmlosfernom phosphine. preparation of amines and their derivatives such as Although the chemistry of organophosphorus α-amino acids. compounds devoted a considerable number of monographs, but the reaction of organic compounds of arsenic, antimony and bismuth are very limited. -

Midas Arsine
® Midas SENSOR CARTRIDGE SPECIFICATIONS Arsine (AsH3), Germane (GeH4) MIDAS-S-ASH, MIDAS-E-ASH Gas Measured Measured Arsine (AsH3) Cross Sensitivities Each Midas® sensor is potentially cross sensitive to other gases Cartridge Part Number MIDAS-S-ASH 1 year standard warranty MIDAS-E-ASH 2 year extended warranty and this may cause a gas reading when exposed to other gases than those originally designated. The table below presents typical Sensor Technology 3 electrode electrochemical cell readings that will be observed when a new sensor cartridge is Measuring Range (ppm) AsH 0 – 0.2ppm exposed to the cross sensitive gas (or a mixture of gases containing 3 the cross sensitive species). Minimum Alarm 1 Set Point 0.025ppm Gas / Vapor Chemical Concentration Reading Repeatability < ± 2% of measured value Formula applied (ppm) (ppm AsH3) Linearity < ± 10% of measured value Ammonia NH3 108 <0.1 Response Time t62.5 < 15 seconds Carbon Dioxide CO2 5,000 0 Sensor Cartridge Life Expectancy ≥ 24 months under typical application conditions Carbon Monoxide CO 85 0 Operating Temperature 0°C to + 40°C (32°C to 104°F) Chlorine CI2 TBD <-0.05 Effect of Temperature Diborane B2H6 0.1 0.05 Zero < ± 0.004 ppm / °C (0° to 40°C) Sensitivity < ± 0.9% of measured value / °C Disilane Si2H6 0.27 0.12 Operating Humidity (continuous) 10 – 95% rH non-condensing Germane GeH4 0.27 0.05 Effect of Humidity Hydrogen H2 3100 <-0.05 Zero Initial short term drift at abrupt rH change (< 0.001 ppm / % rH) Sensitivity < ± 0.2% of measured value / % rH Hydrogen Chloride HCI 7.9 0 -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime.