Armadillo-Repeat Protein Functions: Questions for Little Creatures Tewari, R; Bailes, E; Bunting, KA; Coates, Juliet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -
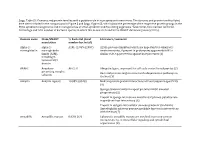
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

CTNNBL1 (NM 030877) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP324836 CTNNBL1 (NM_030877) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human catenin, beta like 1 (CTNNBL1) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 65 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_110517 Locus ID: 56259 UniProt ID: Q8WYA6 RefSeq Size: 1897 Cytogenetics: 20q11.23 RefSeq ORF: 1689 Synonyms: C20orf33; dJ633O20.1; NAP; P14L; PP8304 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 CTNNBL1 (NM_030877) Human Recombinant Protein – TP324836 Summary: The protein encoded by this gene is a component of the pre-mRNA-processing factor 19-cell division cycle 5-like (PRP19-CDC5L) protein complex, which activates pre-mRNA splicing and is an integral part of the spliceosome. The encoded protein is also a nuclear localization sequence binding protein, and binds to activation-induced deaminase and is important for antibody diversification. -

Original Communication Genomic and Expression Array Profiling of Chromosome 20Q Amplicon in Human Colon Cancer Cells
128 Original Communication Genomic and expression array profiling of chromosome 20q amplicon in human colon cancer cells Jennifer l Carter, Li Jin,1 Subrata Sen University of Texas - M. D. Anderson Cancer Center, 1PD, University of Cincinnati sion and metastasis.[1,2] Characterization of genomic BACKGROUND: Gain of the q arm of chromosome 20 in human colorectal cancer has been associated with poorer rearrangements is, therefore, a major area of investiga- survival time and has been reported to increase in frequency tion being pursued by the cancer research community. from adenomas to metastasis. The increasing frequency of chromosome 20q amplification during colorectal cancer Amplification of genomic DNA is one such form of rear- progression and the presence of this amplification in carci- rangement that leads to an increase in the copy num- nomas of other tissue origin has lead us to hypothesize ber of specific genes frequently detected in a variety of that 20q11-13 harbors one or more genes which, when over expressed promote tumor invasion and metastasis. human cancer cell types. Our laboratory has been in- AIMS: Generate genomic and expression profiles of the terested in characterizing amplified genomic regions in 20q amplicon in human cancer cell lines in order to identify genes with increased copy number and expression. cancer cells based on the hypothesis that these seg- MATERIALS AND METHODS: Utilizing genomic sequenc- ments harbor critical genes associated with initiation and/ ing clones and amplification mapping data from our lab or progression of cancer. Gain of chromosome 20q in and other previous studies, BAC/ PAC tiling paths span- ning the 20q amplicon and genomic microarrays were gen- human colorectal cancer has been associated with erated. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Expressed Gene Fusions As Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer
Published OnlineFirst December 14, 2017; DOI: 10.1158/2159-8290.CD-17-0535 RESEARCH ARTICLE Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer Karina J. Matissek1,2, Maristela L. Onozato3, Sheng Sun1,2, Zongli Zheng2,3,4, Andrew Schultz1, Jesse Lee3, Kristofer Patel1, Piiha-Lotta Jerevall2,3, Srinivas Vinod Saladi1,2, Allison Macleay3, Mehrad Tavallai1,2, Tanja Badovinac-Crnjevic5, Carlos Barrios6, Nuran Beşe7, Arlene Chan8, Yanin Chavarri-Guerra9, Marcio Debiasi6, Elif Demirdögen10, Ünal Egeli10, Sahsuvar Gökgöz10, Henry Gomez11, Pedro Liedke6, Ismet Tasdelen10, Sahsine Tolunay10, Gustavo Werutsky6, Jessica St. Louis1, Nora Horick12, Dianne M. Finkelstein2,12, Long Phi Le2,3, Aditya Bardia1,2, Paul E. Goss1,2, Dennis C. Sgroi2,3, A. John Iafrate2,3, and Leif W. Ellisen1,2 ABSTRACT We sought to uncover genetic drivers of hormone receptor–positive (HR+) breast cancer, using a targeted next-generation sequencing approach for detecting expressed gene rearrangements without prior knowledge of the fusion partners. We identified inter- genic fusions involving driver genes, including PIK3CA, AKT3, RAF1, and ESR1, in 14% (24/173) of unselected patients with advanced HR+ breast cancer. FISH confirmed the corresponding chromo- somal rearrangements in both primary and metastatic tumors. Expression of novel kinase fusions in nontransformed cells deregulates phosphoprotein signaling, cell proliferation, and survival in three- dimensional culture, whereas expression in HR+ breast cancer models modulates estrogen-dependent growth and confers hormonal therapy resistance in vitro and in vivo. Strikingly, shorter overall survival was observed in patients with rearrangement-positive versus rearrangement-negative tumors. Cor- respondingly, fusions were uncommon (<5%) among 300 patients presenting with primary HR+ breast cancer. -

Identification of Shared Genetic Susceptibility Locus for Coronary Artery Disease, Type 2 Diabetes and Obesity: a Meta-Analysis of Genome-Wide Studies
Wu et al. Cardiovascular Diabetology 2012, 11:68 CARDIO http://www.cardiab.com/content/11/1/68 VASCULAR DIABETOLOGY REVIEW Open Access Identification of shared genetic susceptibility locus for coronary artery disease, type 2 diabetes and obesity: a meta-analysis of genome-wide studies Chaoneng Wu1,2†, Yunguo Gong1,2†, Jie Yuan1,2, Hui Gong1,2, Yunzeng Zou1,2,3* and Junbo Ge1,2,3* Abstract Type 2 diabetes (2DM), obesity, and coronary artery disease (CAD) are frequently coexisted being as key components of metabolic syndrome. Whether there is shared genetic background underlying these diseases remained unclear. We performed a meta-analysis of 35 genome screens for 2DM, 36 for obesity or body mass index (BMI)-defined obesity, and 21 for CAD using genome search meta-analysis (GSMA), which combines linkage results to identify regions with only weak evidence and provide genetic interactions among different diseases. For each study, 120 genomic bins of approximately 30 cM were defined and ranked according to the best linkage evidence within each bin. For each disease, bin 6.2 achieved genomic significanct evidence, and bin 9.3, 10.5, 16.3 reached suggestive level for 2DM. Bin 11.2 and 16.3, and bin 10.5 and 9.3, reached suggestive evidence for obesity and CAD respectively. In pooled all three diseases, bin 9.3 and 6.5 reached genomic significant and suggestive evidence respectively, being relatively much weaker for 2DM/CAD or 2DM/obesity or CAD/obesity. Further, genomewide significant evidence was observed of bin 16.3 and 4.5 for 2DM/obesity, which is decreased when CAD was added. -

Expression Analysis of Genes Located Within the Common Deleted Region
Leukemia Research 84 (2019) 106175 Contents lists available at ScienceDirect Leukemia Research journal homepage: www.elsevier.com/locate/leukres Research paper Expression analysis of genes located within the common deleted region of del(20q) in patients with myelodysplastic syndromes T ⁎ Masayuki Shiseki , Mayuko Ishii, Michiko Okada, Mari Ohwashi, Yan-Hua Wang, Satoko Osanai, Kentaro Yoshinaga, Naoki Mori, Toshiko Motoji, Junji Tanaka Department of Hematology, Tokyo Women’s Medical University, 8-1 Kawada-cho, Shinjuku-ku, Tokyo, 162-8666, Japan ARTICLE INFO ABSTRACT Keywords: Deletion of the long arm of chromosome 20 (del(20q)) is observed in 5–10% of patients with myelodysplastic Deletion 20q syndromes (MDS). We examined the expression of 28 genes within the common deleted region (CDR) of del Common deleted region (20q), which we previously determined by a CGH array using clinical samples, in 48 MDS patients with (n = 28) Myelodysplastic syndromes or without (n = 20) chromosome 20 abnormalities and control subjects (n = 10). The expression level of 8 of 28 genes was significantly reduced in MDS patients with chromosome 20 abnormalities compared to that of control subjects. In addition, the expression of BCAS4, ADA, and YWHAB genes was significantly reduced in MDS pa- tients without chromosome 20 abnormalities, which suggests that these three genes were commonly involved in the molecular pathogenesis of MDS. To evaluate the clinical significance, we analyzed the impact of the ex- pression level of each gene on overall survival (OS). According to the Cox proportional hazard model, multi- variate analysis indicated that reduced BCAS4 expression was associated with inferior OS, but the difference was not significant (HR, 3.77; 95% CI, 0.995-17.17; P = 0.0509). -

A Systematic, Label-Free Method for Identifying RNA-Associated Proteins in Vivo Provides Insights Into Vertebrate Ciliary Beatin
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.26.966754; this version posted March 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 A systematic, label-free method for identifying RNA- associated proteins in vivo provides insights into vertebrate ciliary beating Kevin Drew*, Chanjae Lee*, Rachael M. Cox, Vy Dang, Caitlin C. Devitt, Ophelia Papoulas, Ryan L. Huizar, Edward M. Marcotte** and John B. Wallingford** Dept. of Molecular Biosciences, Center for Systems and Synthetic Biology, University of Texas, Austin, TX 78712 *These authors contributed equally **To whom correspondence should be addressed: John Wallingford Patterson Labs 2401 Speedway Austin, Texas 78712 [email protected] 512-232-2784 Edward Marcotte 2500 Speedway, MBB 3.148BA Austin, Texas 78712 [email protected] 512-471-5435 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.26.966754; this version posted March 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 2 Abstract: Cell-type specific RNA-associated proteins (RAPs) are essential for development and homeostasis in animals. Despite a massive recent effort to systematically identify RAPs, we currently have few comprehensive rosters of cell-type specific RAPs in vertebrate tissues. Here, we demonstrate the feasibility of determining the RNA-interacting proteome of a defined vertebrate embryonic tissue using DIF-FRAC, a systematic and universal (i.e., label-free) method. -

Armc5 Deletion Causes Developmental Defects and Compromises T-Cell Immune Responses
ARTICLE Received 26 Jan 2016 | Accepted 4 Nov 2016 | Published 7 Feb 2017 DOI: 10.1038/ncomms13834 OPEN Armc5 deletion causes developmental defects and compromises T-cell immune responses Yan Hu 1,*, Linjiang Lao1,*, Jianning Mao1, Wei Jin1, Hongyu Luo1, Tania Charpentier2, Shijie Qi1, Junzheng Peng1, Bing Hu3, Mieczyslaw Martin Marcinkiewicz4, Alain Lamarre2 & Jiangping Wu1,5 Armadillo repeat containing 5 (ARMC5) is a cytosolic protein with no enzymatic activities. Little is known about its function and mechanisms of action, except that gene mutations are associated with risks of primary macronodular adrenal gland hyperplasia. Here we map Armc5 expression by in situ hybridization, and generate Armc5 knockout mice, which are small in body size. Armc5 knockout mice have compromised T-cell proliferation and differentiation into Th1 and Th17 cells, increased T-cell apoptosis, reduced severity of experimental autoimmune encephalitis, and defective immune responses to lymphocytic choriomeningitis virus infection. These mice also develop adrenal gland hyperplasia in old age. Yeast 2-hybrid assays identify 16 ARMC5-binding partners. Together these data indicate that ARMC5 is crucial in fetal development, T-cell function and adrenal gland growth homeostasis, and that the functions of ARMC5 probably depend on interaction with multiple signalling pathways. 1 Centre de recherche (CR), Centre hospitalier de l’Universite´ de Montre´al (CHUM), 900 Rue Saint Denis, Montre´al, Que´bec, Canada H2X 0A9. 2 Institut national de la recherche scientifique—Institut Armand-Frappier (INRS-IAF), 531 Boul. des Prairies, Laval, Que´bec, Canada H7V 1B7. 3 Anatomic Pathology, AmeriPath Central Florida, 4225 Fowler Ave. Tampa, Orlando, Florida 33617, USA. -

Newly Identified Gon4l/Udu-Interacting Proteins
www.nature.com/scientificreports OPEN Newly identifed Gon4l/ Udu‑interacting proteins implicate novel functions Su‑Mei Tsai1, Kuo‑Chang Chu1 & Yun‑Jin Jiang1,2,3,4,5* Mutations of the Gon4l/udu gene in diferent organisms give rise to diverse phenotypes. Although the efects of Gon4l/Udu in transcriptional regulation have been demonstrated, they cannot solely explain the observed characteristics among species. To further understand the function of Gon4l/Udu, we used yeast two‑hybrid (Y2H) screening to identify interacting proteins in zebrafsh and mouse systems, confrmed the interactions by co‑immunoprecipitation assay, and found four novel Gon4l‑interacting proteins: BRCA1 associated protein‑1 (Bap1), DNA methyltransferase 1 (Dnmt1), Tho complex 1 (Thoc1, also known as Tho1 or HPR1), and Cryptochrome circadian regulator 3a (Cry3a). Furthermore, all known Gon4l/Udu‑interacting proteins—as found in this study, in previous reports, and in online resources—were investigated by Phenotype Enrichment Analysis. The most enriched phenotypes identifed include increased embryonic tissue cell apoptosis, embryonic lethality, increased T cell derived lymphoma incidence, decreased cell proliferation, chromosome instability, and abnormal dopamine level, characteristics that largely resemble those observed in reported Gon4l/udu mutant animals. Similar to the expression pattern of udu, those of bap1, dnmt1, thoc1, and cry3a are also found in the brain region and other tissues. Thus, these fndings indicate novel mechanisms of Gon4l/ Udu in regulating CpG methylation, histone expression/modifcation, DNA repair/genomic stability, and RNA binding/processing/export. Gon4l is a nuclear protein conserved among species. Animal models from invertebrates to vertebrates have shown that the protein Gon4-like (Gon4l) is essential for regulating cell proliferation and diferentiation.