PRDM9 in Der Primatenevolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

LETÍCIA SIGNORI DE CASTRO Estudo Do Perfil Proteico E
LETÍCIA SIGNORI DE CASTRO Estudo do perfil proteico e epigenético do núcleo espermático bovino com influência na produção in vitro de embriões São Paulo 2018 LETÍCIA SIGNORI DE CASTRO Estudo do perfil proteico e epigenético do núcleo espermático bovino com influência na produção in vitro de embriões Tese apresentada ao Programa de Pós- Graduação em Reprodução Animal da Faculdade de Medicina Veterinária e Zootecnia da Universidade de São Paulo para obtenção do título de Doutor em Ciências Departamento: Reprodução Animal Área de concentração: Reprodução Animal Orientadora: Profa. Dra. Mayra Elena Ortiz D’Ávila Assumpção São Paulo 2018 Autorizo a reprodução parcial ou total desta obra, para fins acadêmicos, desde que citada a fonte. DADOS INTERNACIONAIS DE CATALOGAÇÃO NA PUBLICAÇÃO (Biblioteca Virginie Buff D’Ápice da Faculdade de Medicina Veterinária e Zootecnia da Universidade de São Paulo) T. 3727 Castro, Letícia Signori de FMVZ Estudo do perfil proteico e epigenético do núcleo espermático bovino com influência na produção in vitro de embriões / Letícia Signori de Castro. – 2018. 98 f. : il. Tese (Doutorado) – Universidade de São Paulo. Faculdade de Medicina Veterinária e Zootecnia. Departamento de Reprodução Animal, São Paulo, 2018. Programa de Pós-Graduação: Reprodução Animal. Área de concentração: Reprodução Animal. Orientadora: Profa. Dra. Mayra Elena Ortiz D'Ávila Assumpção. 1. Espermatozoide. 2. Fertilidade. 3. Proteômica. 4. Epigenética. I. Título. Ficha catalográfica elaborada pela bibliotecária Maria Aparecida Laet, CRB 5673-8, da FMVZ/USP. FOLHA DE AVALIAÇÃO Nome: CASTRO, Letícia Signori de Título: Estudo do perfil proteico e epigenético do núcleo espermático bovino com influência na produção in vitro de embriões Tese apresentada ao Programa de Pós- Graduação em Reprodução Animal da Faculdade de Medicina Veterinária e Zootecnia da Universidade de São Paulo para obtenção do título de Doutor em Ciências Data: ____ / ____ / _____ Banca examinadora Prof. -

Functional Annotation of the Human Retinal Pigment Epithelium
BMC Genomics BioMed Central Research article Open Access Functional annotation of the human retinal pigment epithelium transcriptome Judith C Booij1, Simone van Soest1, Sigrid MA Swagemakers2,3, Anke HW Essing1, Annemieke JMH Verkerk2, Peter J van der Spek2, Theo GMF Gorgels1 and Arthur AB Bergen*1,4 Address: 1Department of Molecular Ophthalmogenetics, Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences (KNAW), Meibergdreef 47, 1105 BA Amsterdam, the Netherlands (NL), 2Department of Bioinformatics, Erasmus Medical Center, 3015 GE Rotterdam, the Netherlands, 3Department of Genetics, Erasmus Medical Center, 3015 GE Rotterdam, the Netherlands and 4Department of Clinical Genetics, Academic Medical Centre Amsterdam, the Netherlands Email: Judith C Booij - [email protected]; Simone van Soest - [email protected]; Sigrid MA Swagemakers - [email protected]; Anke HW Essing - [email protected]; Annemieke JMH Verkerk - [email protected]; Peter J van der Spek - [email protected]; Theo GMF Gorgels - [email protected]; Arthur AB Bergen* - [email protected] * Corresponding author Published: 20 April 2009 Received: 10 July 2008 Accepted: 20 April 2009 BMC Genomics 2009, 10:164 doi:10.1186/1471-2164-10-164 This article is available from: http://www.biomedcentral.com/1471-2164/10/164 © 2009 Booij et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

WO 2013/184908 A2 12 December 2013 (12.12.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2013/184908 A2 12 December 2013 (12.12.2013) P O P C T (51) International Patent Classification: Jr.; One Procter & Gamble Plaza, Cincinnati, Ohio 45202 G06F 19/00 (201 1.01) (US). HOWARD, Brian, Wilson; One Procter & Gamble Plaza, Cincinnati, Ohio 45202 (US). (21) International Application Number: PCT/US20 13/044497 (74) Agents: GUFFEY, Timothy, B. et al; c/o The Procter & Gamble Company, Global Patent Services, 299 East 6th (22) Date: International Filing Street, Sycamore Building, 4th Floor, Cincinnati, Ohio 6 June 2013 (06.06.2013) 45202 (US). (25) Filing Language: English (81) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (30) Priority Data: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 61/656,218 6 June 2012 (06.06.2012) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (71) Applicant: THE PROCTER & GAMBLE COMPANY HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, [US/US]; One Procter & Gamble Plaza, Cincinnati, Ohio KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 45202 (US). MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, (72) Inventors: XU, Jun; One Procter & Gamble Plaza, Cincin SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, nati, Ohio 45202 (US). -

Genetic Epidemiology of Atopy and Asthma
Genetic Epidemiology of Atopy and Asthma By Yize I Wan, BMedsci. (Hons) Thesis submitted to the University of Nottingham for the degree of Doctor of Philosophy May 2011 To my family, with all my love Contents ABSTRACT ..........................................................................................................................I ACKNOWLEDGEMENTS ................................................................................................ II ABBREVIATIONS ............................................................................................................III PUBLICATIONS RELEVANT TO THESIS .................................................................... IV CHAPTER 1 ........................................................................................................................ 1 GENERAL INTRODUCTION ............................................................................................ 1 1.1 ALLERGIC DISEASE ................................................................................................... 1 1.1.1 Definition ............................................................................................................. 1 1.1.2 Epidemiology ....................................................................................................... 1 1.1.3 Pathogenesis ........................................................................................................ 1 1.1.4 Allergens ............................................................................................................. -

Thesis Was Conducted at the Netherlands Institute for Neuro- Science
UvA-DARE (Digital Academic Repository) Function and pathology of the human retinal pigment epithelium Booij, J.C. Publication date 2010 Document Version Final published version Link to publication Citation for published version (APA): Booij, J. C. (2010). Function and pathology of the human retinal pigment epithelium. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:04 Oct 2021 Function and pathology of the human retinal pigment epithelium Function and pathology of the human retinal pigment epithelium Judith C. Booij Judith C. Booij Booij_Omslag.indd 1 18-06-10 10:24 Function and pathology of the human retinal pigment epithelium Function and pathology of the human retinal pigment epithelium ACADEMISCH PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Universiteit van Amsterdam op gezag van de Rector Magnificus Mw. -

Susceptible Gene of Stasis-Stagnation Constitution from Genome-Wide
Huang et al. BMC Complementary and Alternative Medicine (2015) 15:229 DOI 10.1186/s12906-015-0761-x RESEARCH ARTICLE Open Access Susceptible gene of stasis-stagnation constitution from genome-wide association study related to cardiovascular disturbance and possible regulated traditional Chinese medicine Kuo-Chin Huang1,2,4, Hung-Jin Huang3, Ching-Chu Chen4,5, Chwen-Tzuei Chang4,5,Tzu-YuanWang4,5, Rong-Hsing Chen4,5, Yu-Chian Chen6,7,8* and Fuu-Jen Tsai4,6,9* Abstract Background: This study identified susceptible loci related to the Yu-Zhi (YZ) constitution, which indicates stasis-stagnation, found in a genome-wide association study (GWAS) in patients with type 2 diabetes and possible regulated traditional Chinese medicine (TCM) using docking and molecular dynamics (MD) simulation. Methods: Non-aboriginal Taiwanese with type 2 diabetes were recruited. Components of the YZ constitution were assessed by a self-reported questionnaire. Genome-wide SNP genotypes were obtained using the Illumina HumanHap550 platform. The world’s largest TCM database (http://tcm.cmu.edu.tw/) was employed to investigate potential compounds for PON2 interactions. Results: The study involved 1,021 unrelated individuals with type 2 diabetes. Genotyping data were obtained from 947 of the 1,021 participants. The GWAS identified 22 susceptible single nucleotide polymorphisms on 13 regions of 11 chromosomes for the YZ constitution. Genotypic distribution showed that PON2 on chromosome 7 was most significantly associated with the risk of the YZ constitution. Docking and MD simulation indicated 13-hydroxy-(9E_11E)-octadecadienoic acid was the most stable TCM ligand. Conclusions: Risk loci occurred in PON2, which has antioxidant properties that might protect against atherosclerosis and hyperglycemia, showing it is a susceptible gene for the YZ constitution and possible regulation by 13-hydroxy-(9E_11E)-octadecadienoic acid. -

SUPPLEMENTARY APPENDIX a B-Cell Receptor-Related Gene Signature Predicts Response to Ibrutinib Treatment in Mantle Cell Lymphoma Cell Lines
SUPPLEMENTARY APPENDIX A B-cell receptor-related gene signature predicts response to ibrutinib treatment in mantle cell lymphoma cell lines Tiziana D'Agaro, 1 Antonella Zucchetto, 1 Filippo Vit, 1,2 Tamara Bittolo, 1 Erika Tissino, 1 Francesca Maria Rossi, 1 Massimo Degan, 1 Francesco Zaja, 3 Pietro Bulian, 1 Michele Dal Bo, 1 Simone Ferrero, 4,5 Marco Ladetto, 4,6 Alberto Zamò, 7 Valter Gattei 1* and Riccardo Bomben 1* *VG and RB equally contributed to this work as senior authors 1Clinical and Experimental Onco-Hematology Unit, Centro di Riferimento Oncologico di Aviano (CRO), IRCCS, Aviano; 2Department of Life Science, Univer - sity of Trieste, Trieste; 3Department of Internal Medicine and Haematology, Maggiore General Hospital, University of Trieste, Trieste; 4Department of Molecular Biotechnologies and Health Sciences, Hematology Division 1, University of Torino, Torino; 5Hematology Division 1, AOU “Città della Salute e della Scienza di Torino” University-Hospital, Torino; 6SC Ematologia Azienda Ospedaliera Nazionale SS. Antonio e Biagio e Cesare Arrigo, Alessandria and 7Department of Oncology, University of Torino, Torino, Italy Correspondence: RICCARDO BOMBEN - [email protected] doi:10.3324/haematol.2018.212811 A B-cell receptor-related gene signature predicts response to ibrutinib treatmeant in Mantle Cell Lymphoma cell lines Supplemental Information: • Supplemental Materials and Methods • Supplemental Figure: o Figure S1. Anti-IgM stimulation and ibrutinib treatment on primary MCL samples. • Supplemental Tables: o Table S1. Differentially expressed genes between BCR low and BCR high MCL cell lines. o Table S2. TP53 mutational status in MCL cell line models 1 MCL cell lines Six different MCL cell line models (Rec-1, Jeko-1, Mino, JVM-2, Granta-519, and Z-138)1 were used for GEP studies and functional experiments. -

Universidade De Lisboa Faculdade De Ciências Departamento De Química E Bioquímica
Universidade de Lisboa Faculdade de Ciências Departamento de Química e Bioquímica Molecular characterisation of the poorly differentiated and undifferentiated thyroid carcinomas using genome-wide approaches Jaime Miguel Gomes Pita Doutoramento em Bioquímica Especialidade em Genética Molecular 2013 Universidade de Lisboa Faculdade de Ciências Departamento de Química e Bioquímica Molecular characterisation of the poorly differentiated and undifferentiated thyroid carcinomas using genome-wide approaches Jaime Miguel Gomes Pita Tese orientada pelo Professor Doutor Valeriano Alberto Leite (Instituto Português de Oncologia de Lisboa, Francisco Gentil) e Professora Doutora Maria Luísa Cyrne (Faculdade de Ciências da Universidade de Lisboa) especialmente elaborada para a obtenção do grau de doutor em Bioquímica, especialidade em Genética Molecular 2013 A presente dissertação foi redigida de acordo com o disposto no artigo 45.º do Despacho n.º 4624/2012 do Diário da República 2.ª série - N.º 65 - 30 de Março de 2012 * * * As opiniões expressas nesta publicação são da exclusiva responsabilidade do seu autor. (...) (...) Eles não sabem que o sonho Eles não sabem, nem sonham, é vinho, é espuma, é fermento, que o sonho comanda a vida. bichinho álacre e sedento, Que sempre que um homem sonha de focinho pontiagudo, o mundo pula e avança que fossa através de tudo como bola colorida num perpétuo movimento. entre as mãos de uma criança. António Gedeão, Pedra Filosofal in ‘Movimento Perpétuo’ (1956) ‘The deeper we search the more we find there is to know, and as long as human life exists, I believe it will always be so.’ Albert Einstein Agradecimentos Agradecimentos Na presente tese de doutoramento são apresentados os resultados do trabalho de investigação realizado no Grupo de Endocrinologia Molecular da Unidade de Investigação de Patobiologia Molecular (UIPM) no Instituto Português de Oncologia de Lisboa, Francisco Gentil (IPOLFG) entre Janeiro de 2009 e Dezembro de 2012, sob a orientação do Professor Doutor Valeriano Alberto Leite e co-orientação da Doutora Branca Maria Cavaco. -
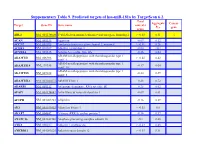
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Wan, Yize Isalina (2011) Genetic Epidemiology of Atopy and Asthma
Wan, Yize Isalina (2011) Genetic epidemiology of atopy and asthma. PhD thesis, University of Nottingham. Access from the University of Nottingham repository: http://eprints.nottingham.ac.uk/12224/1/Print_YWan_ThesisFinal_Oct2011.pdf Copyright and reuse: The Nottingham ePrints service makes this work by researchers of the University of Nottingham available open access under the following conditions. · Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. · To the extent reasonable and practicable the material made available in Nottingham ePrints has been checked for eligibility before being made available. · Copies of full items can be used for personal research or study, educational, or not- for-profit purposes without prior permission or charge provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. · Quotations or similar reproductions must be sufficiently acknowledged. Please see our full end user licence at: http://eprints.nottingham.ac.uk/end_user_agreement.pdf A note on versions: The version presented here may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the repository url above for details on accessing the published version and note that access may require a subscription. For more information, please contact [email protected] Genetic Epidemiology of Atopy and Asthma By Yize I Wan, BMedsci. (Hons) Thesis submitted to the University of Nottingham for the degree of Doctor of Philosophy May 2011 To my family, with all my love Contents ABSTRACT ..........................................................................................................................I ACKNOWLEDGEMENTS ............................................................................................... -

Analyse Von Herkunft Und Funktioneller Aktivität Humaner Membranständiger Glucocorticoidrezeptoren
Analyse von Herkunft und funktioneller Aktivität humaner membranständiger Glucocorticoidrezeptoren vorgelegt von Diplom-Biochemikerin Cindy Strehl aus Berlin von der Fakultät III – Prozesswissenschaften der Technischen Universität Berlin zur Erlangung des akademischen Grades Doktorin der Naturwissenschaften - Dr. rer. nat. - genehmigte Dissertation Promotionsausschuss: Vorsitzender: Prof. Dr. Peter Neubauer Berichter: Prof. Dr. Roland Lauster Berichter: Prof. Dr. Frank Buttgereit Tag der wissenschaftlichen Aussprache: 24. Oktober 2011 Berlin 2011 D83 2 Meiner Familie... 3 4 „Wer A sagt, der muss nicht B sagen. Er kann auch erkennen, dass A falsch war.“ (Berthold Brecht 1898 – 1956) Der Jasager. Der Neinsager 1929 - 1930 5 Inhaltsverzeichnis 6 Inhaltsverzeichnis IInnhhaallttssvveerrzzeeiicchhnniiss 7 Inhaltsverzeichnis 8 Inhaltsverzeichnis Inhaltsverzeichnis Zusammenfassung........................................................................ 17 Abstract ....................................................................................... 19 1. Einleitung ................................................................................ 23 1.1 Hormone – die Botenstoffe des Körpers .................................................................................. 23 1.1.1 Allgemeines ....................................................................................................................... 23 1.1.2 Die Steroidhormone: Ein Grundgerüst - Unterschiedliche Wirkungen ............................ 24 1.1.2.1 Biosynthese und Regulation -

Lyl1 Interacts with CREB1 and Alters Expression of CREB1 Target Genes☆ ⁎ Serban San-Marina A, Youqi Han A, Fernando Suarez Saiz A, Michael R
Available online at www.sciencedirect.com Biochimica et Biophysica Acta 1783 (2008) 503–517 www.elsevier.com/locate/bbamcr Lyl1 interacts with CREB1 and alters expression of CREB1 target genes☆ ⁎ Serban San-Marina a, YouQi Han a, Fernando Suarez Saiz a, Michael R. Trus b,1, Mark D. Minden a, a Ontario Cancer Institute/Princess Margaret Hospital, 610 University Avenue 9-111, Toronto, Ontario, Canada M5G 2M9 b Pathology and Molecular Medicine, Henderson General Hospital, Hamilton, Ontario, ON L8V 1A1, Canada Received 27 July 2007; received in revised form 19 November 2007; accepted 20 November 2007 Available online 26 December 2007 Abstract The basic helix-loop-helix (bHLH) transcription factor family contains key regulators of cellular proliferation and differentiation as well as the suspected oncoproteins Tal1 and Lyl1. Tal1 and Lyl1 are aberrantly over-expressed in leukemia as a result of chromosomal translocations, or other genetic or epigenetic events. Protein-protein and protein-DNA interactions described so far are mediated by their highly homologous bHLH domains, while little is known about the function of other protein domains. Hetero-dimers of Tal1 and Lyl1 with E2A or HEB, decrease the rate of E2A or HEB homo-dimer formation and are poor activators of transcription. In vitro, these hetero-dimers also recognize different binding sites from homo-dimer complexes, which may also lead to inappropriate activation or repression of promoters in vivo. Both mechanisms are thought to contribute to the oncogenic potential of Tal1 and Lyl1. Despite their bHLH structural similarity, accumulating evidence suggests that Tal1 and Lyl1 target different genes. This raises the possibility that domains flanking the bHLH region, which are distinct in the two proteins, may participate in target recognition.