Phenotype Expression Variability in Children with GABRB3 Heterozygous Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Material
Supplementary Material Table S1: Significant downregulated KEGGs pathways identified by DAVID following exposure to five cinnamon- based phenylpropanoids (p < 0.05). p-value Term: Genes (Benjamini) Cytokine-cytokine receptor interaction: FASLG, TNFSF14, CXCL11, IL11, FLT3LG, CCL3L1, CCL3L3, CXCR6, XCR1, 2.43 × 105 RTEL1, CSF2RA, TNFRSF17, TNFRSF14, CCNL2, VEGFB, AMH, TNFRSF10B, INHBE, IFNB1, CCR3, VEGFA, CCR2, IL12A, CCL1, CCL3, CXCL5, TNFRSF25, CCR1, CSF1, CX3CL1, CCL7, CCL24, TNFRSF1B, IL12RB1, CCL21, FIGF, EPO, IL4, IL18R1, FLT1, TGFBR1, EDA2R, HGF, TNFSF8, KDR, LEP, GH2, CCL13, EPOR, XCL1, IFNA16, XCL2 Neuroactive ligand-receptor interaction: OPRM1, THRA, GRIK1, DRD2, GRIK2, TACR2, TACR1, GABRB1, LPAR4, 9.68 × 105 GRIK5, FPR1, PRSS1, GNRHR, FPR2, EDNRA, AGTR2, LTB4R, PRSS2, CNR1, S1PR4, CALCRL, TAAR5, GABRE, PTGER1, GABRG3, C5AR1, PTGER3, PTGER4, GABRA6, GABRA5, GRM1, PLG, LEP, CRHR1, GH2, GRM3, SSTR2, Chlorogenic acid Chlorogenic CHRM3, GRIA1, MC2R, P2RX2, TBXA2R, GHSR, HTR2C, TSHR, LHB, GLP1R, OPRD1 Hematopoietic cell lineage: IL4, CR1, CD8B, CSF1, FCER2, GYPA, ITGA2, IL11, GP9, FLT3LG, CD38, CD19, DNTT, 9.29 × 104 GP1BB, CD22, EPOR, CSF2RA, CD14, THPO, EPO, HLA-DRA, ITGA2B Cytokine-cytokine receptor interaction: IL6ST, IL21R, IL19, TNFSF15, CXCR3, IL15, CXCL11, TGFB1, IL11, FLT3LG, CXCL10, CCR10, XCR1, RTEL1, CSF2RA, IL21, CCNL2, VEGFB, CCR8, AMH, TNFRSF10C, IFNB1, PDGFRA, EDA, CXCL5, TNFRSF25, CSF1, IFNW1, CNTFR, CX3CL1, CCL5, TNFRSF4, CCL4, CCL27, CCL24, CCL25, CCL23, IFNA6, IFNA5, FIGF, EPO, AMHR2, IL2RA, FLT4, TGFBR2, EDA2R, -

Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed Hipscs To
Hindawi Publishing Corporation Stem Cells International Volume 2013, Article ID 784629, 25 pages http://dx.doi.org/10.1155/2013/784629 Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed hiPSCs to Differentiated Neuronal and Cardiac Progeny Leonhard Linta,1 Marianne Stockmann,1 Qiong Lin,2 André Lechel,3 Christian Proepper,1 Tobias M. Boeckers,1 Alexander Kleger,3 and Stefan Liebau1 1 InstituteforAnatomyCellBiology,UlmUniversity,Albert-EinsteinAllee11,89081Ulm,Germany 2 Institute for Biomedical Engineering, Department of Cell Biology, RWTH Aachen, Pauwelstrasse 30, 52074 Aachen, Germany 3 Department of Internal Medicine I, Ulm University, Albert-Einstein Allee 11, 89081 Ulm, Germany Correspondence should be addressed to Alexander Kleger; [email protected] and Stefan Liebau; [email protected] Received 31 January 2013; Accepted 6 March 2013 Academic Editor: Michael Levin Copyright © 2013 Leonhard Linta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Ion channels are involved in a large variety of cellular processes including stem cell differentiation. Numerous families of ion channels are present in the organism which can be distinguished by means of, for example, ion selectivity, gating mechanism, composition, or cell biological function. To characterize the distinct expression of this group of ion channels we have compared the mRNA expression levels of ion channel genes between human keratinocyte-derived induced pluripotent stem cells (hiPSCs) and their somatic cell source, keratinocytes from plucked human hair. This comparison revealed that 26% of the analyzed probes showed an upregulation of ion channels in hiPSCs while just 6% were downregulated. -

Identification of Key Genes and Pathways Involved in Response To
Deng et al. Biol Res (2018) 51:25 https://doi.org/10.1186/s40659-018-0174-7 Biological Research RESEARCH ARTICLE Open Access Identifcation of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing Xiuling Deng1,2†, Dong Wang3†, Shenyuan Wang1, Haisheng Wang2 and Huanmin Zhou1* Abstract Purpose: This aim of this study was to investigate the key genes and pathways involved in the response to pain in goat and sheep by transcriptome sequencing. Methods: Chronic pain was induced with the injection of the complete Freund’s adjuvant (CFA) in sheep and goats. The animals were divided into four groups: CFA-treated sheep, control sheep, CFA-treated goat, and control goat groups (n 3 in each group). The dorsal root ganglions of these animals were isolated and used for the construction of a cDNA= library and transcriptome sequencing. Diferentially expressed genes (DEGs) were identifed in CFA-induced sheep and goats and gene ontology (GO) enrichment analysis was performed. Results: In total, 1748 and 2441 DEGs were identifed in CFA-treated goat and sheep, respectively. The DEGs identi- fed in CFA-treated goats, such as C-C motif chemokine ligand 27 (CCL27), glutamate receptor 2 (GRIA2), and sodium voltage-gated channel alpha subunit 3 (SCN3A), were mainly enriched in GO functions associated with N-methyl- D-aspartate (NMDA) receptor, infammatory response, and immune response. The DEGs identifed in CFA-treated sheep, such as gamma-aminobutyric acid (GABA)-related DEGs (gamma-aminobutyric acid type A receptor gamma 3 subunit [GABRG3], GABRB2, and GABRB1), SCN9A, and transient receptor potential cation channel subfamily V member 1 (TRPV1), were mainly enriched in GO functions related to neuroactive ligand-receptor interaction, NMDA receptor, and defense response. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -
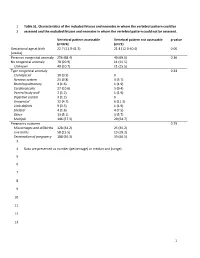
1 Table S1. Characteristics of the Included Fetuses and Neonates in Whom the Vertebral Pattern Could Be 1 Assessed and the Exclu
1 Table S1. Characteristics of the included fetuses and neonates in whom the vertebral pattern could be 2 assessed and the excluded fetuses and neonates in whom the vertebral pattern could not be assessed. Vertebral pattern assessable Vertebral pattern not assessable p-value (n=374) (n=71) Gestational age at birth 22.7 (11.9-41.3) 21.4 (12.0-40.4) 0.06 (weeks) Presence congenital anomaly 256 (68.4) 49 (69.0) 0.36 No congenital anomaly 78 (20.9) 11 (15.5) Unknown 40 (10.7) 11 (15.5) Type congenital anomaly 0.24 Craniofacial 10 (3.9) 0 Nervous system 25 (9.8) 3 (5.7) Bronchopulmonary 4 (1.6) 1 (1.9) Cardiovascular 27 (10.6) 5 (9.4) Ventral body wall 3 (1.2) 1 (1.9) Digestive system 3 (1.2) 0 Urogenital 12 (4.7) 6 (11.3) Limb defects 9 (3.5) 1 (1.9) Skeletal 4 (1.6) 4 (7.5) Other 13 (5.1) 3 (5.7) Multiple 146 (57.3) 29 (54.7) Pregnancy outcome 0.79 Miscarriages and stillbirths 128 (34.2) 25 (35.2) Live births 58 (15.5) 13 (18.3) Termination of pregnancy 188 (50.3) 33 (46.5) 3 4 Data are presented as number (percentage) or median and (range). 5 6 7 8 9 10 11 12 13 1 14 Table S2. Gestational age at birth, age at death, presence of congenital abnormalities and cause of death of the included live births. GA at birth Age at death Cervical Congenital abnormalities Cause of death (weeks) (days) rib(s) 22.3 0 No. -

Replicated Risk Nicotinic Cholinergic Receptor Genes for Nicotine Dependence
G C A T T A C G G C A T genes Article Replicated Risk Nicotinic Cholinergic Receptor Genes for Nicotine Dependence Lingjun Zuo 1, Rolando Garcia-Milian 2, Xiaoyun Guo 1,3,4,*, Chunlong Zhong 5,*, Yunlong Tan 6, Zhiren Wang 6, Jijun Wang 3, Xiaoping Wang 7, Longli Kang 8, Lu Lu 9,10, Xiangning Chen 11,12, Chiang-Shan R. Li 1 and Xingguang Luo 1,6,* 1 Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA; [email protected] (L.Z.); [email protected] (C.-S.R.L.) 2 Curriculum & Research Support Department, Cushing/Whitney Medical Library, Yale University School of Medicine, New Haven, CT 06510, USA; [email protected] 3 Shanghai Mental Health Center, Shanghai 200030, China; [email protected] 4 Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Haven, CT 06510, USA 5 Department of Neurosurgery, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China 6 Biological Psychiatry Research Center, Beijing Huilongguan Hospital, Beijing 100096, China; [email protected] (Y.T.); [email protected] (Z.W.) 7 Department of Neurology, Shanghai First People’s Hospital, Shanghai Jiao Tong University, Shanghai 200080, China; [email protected] 8 Key Laboratory for Molecular Genetic Mechanisms and Intervention Research on High Altitude Diseases of Tibet Autonomous Region, Xizang Minzu University School of Medicine, Xianyang, Shanxi 712082, China; [email protected] 9 Provincial Key Laboratory for Inflammation and Molecular Drug Target, Medical -

Potential Role of Genomic Imprinted Genes and Brain Developmental
Li et al. BMC Medical Genomics (2020) 13:54 https://doi.org/10.1186/s12920-020-0693-2 RESEARCH ARTICLE Open Access Potential role of genomic imprinted genes and brain developmental related genes in autism Jian Li1*† , Xue Lin2†, Mingya Wang1†,YunyunHu1, Kaiyu Xue1,ShuanglinGu1,LiLv1, Saijun Huang3 and Wei Xie1* Abstract Background: Autism is a complex disease involving both environmental and genetic factors. Recent efforts have implicated the correlation of genomic imprinting and brain development in autism, however the pathogenesis of autism is not completely clear. Here, we used bioinformatic tools to provide a comprehensive analysis of the autism-related genes, genomic imprinted genes and the spatially and temporally differentially expressed genes of human brain, aiming to explore the relationship between autism, brain development and genomic imprinting. Methods: This study analyzed the distribution correlation between autism-related genes and imprinted genes on chromosomes using sliding windows and statistical methods. The normal brains’ gene expression microarray data were reanalyzed to construct a spatio-temporal coordinate system of gene expression during brain development. Finally, we intersected the autism-related genes, imprinted genes and brain spatio-temporally differentially expressed genes for further analysis to find the major biological processes that these genes involved. Results: We found a positive correlation between the autism-related genes’ and imprinted genes’ distribution on chromosomes. Through the analysis of the normal brain microarray data, we constructed a spatio-temporal coordinate system of gene expression during human brain development, and obtained 13 genes that are differentially expressed in the process of brain development, which are both autism-related genes and imprinted genes. -

Distinct Diagnostic and Prognostic Values of Γ‑Aminobutyric Acid Type a Receptor Family Genes in Patients with Colon Adenocarcinoma
ONCOLOGY LETTERS 20: 275-291, 2020 Distinct diagnostic and prognostic values of γ‑aminobutyric acid type A receptor family genes in patients with colon adenocarcinoma LING YAN1, YI‑ZHEN GONG1, MENG‑NAN SHAO2, GUO‑TIAN RUAN1, HAI‑LUN XIE1, XI‑WEN LIAO3, XIANG‑KUN WANG3, QUAN‑FA HAN3, XIN ZHOU3, LI‑CHENG ZHU4, FENG GAO1 and JIA‑LIANG GAN1 1Department of Colorectal and Anal Surgery, The First Affiliated Hospital of Guangxi Medical University; 2Life Sciences Institute, Guangxi Medical University; 3Department of Hepatobiliary Surgery, The First Affiliated Hospital of Guangxi Medical University; 4Department of Immunology, School of Preclinical Medicine, Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region 530021, P.R. China Received July 11, 2019; Accepted February 7, 2020 DOI: 10.3892/ol.2020.11573 Abstract. In the present study, the significance of GABAA of cell matrix adhesion, integrin binding, angiogenesis, endo- genes in colon adenocarcinoma (COAD) were investigated thelial growth factor and endothelial migration regulation in from the view of diagnosis and prognosis. All data were patients with COAD with GABRD overexpression. GABRB1, achieved from The Cancer Genome Atlas. Overall survival GABRD, GABRP and GABRQ were associated with the was analyzed by the Kaplan‑Meier analyses and Cox prognostic factors of COAD. The expression levels of regression model and the hazard ratios and 95% confidence GABRA2, GABRA3, GABRB2, GABRB3, GABRG2, GABRD interval were calculated for computation. The Database for and GABRE may allow differentiation between tumor tissues Annotation, Visualization and Integrated Discovery, and the and adjacent normal tissues. Biological Networks Gene Ontology (BiNGO) softwares were applied to assess the biological processes and Kyoto Introduction Encyclopedia of Genes and Genomes (KEGG) was used for pathway analysis to predict the biological function of GABAA Colorectal cancer (CRC) is a type of malignant tumor origi- genes. -

GABRG3 Antibody (N-Term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap4800a
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 GABRG3 Antibody (N-term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP4800a Specification GABRG3 Antibody (N-term) - Product Information Application WB, IHC-P, FC,E Primary Accession Q99928 Reactivity Human, Mouse Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Calculated MW 54289 Antigen Region 1-29 GABRG3 Antibody (N-term) - Additional Information Gene ID 2567 Western blot analysis of GABRG3 Antibody (N-term) (Cat. #AP4800a) in mouse brain Other Names tissue lysates (35ug/lane). GABRG3 (arrow) Gamma-aminobutyric acid receptor subunit gamma-3, GABA(A) receptor subunit was detected using the purified Pab. gamma-3, GABRG3 Target/Specificity This GABRG3 antibody is generated from rabbits immunized with a KLH conjugated synthetic peptide between 1-29 amino acids from the N-terminal region of human GABRG3. Dilution WB~~1:1000 IHC-P~~1:50~100 FC~~1:10~50 Format GABRG3 Antibody (N-term) (Cat. #AP4800a) Purified polyclonal antibody supplied in PBS IHC analysis in formalin fixed and paraffin with 0.09% (W/V) sodium azide. This embedded brain tissue followed by antibody is purified through a protein A peroxidase conjugation of the secondary column, followed by peptide affinity antibody and DAB staining. This data purification. demonstrates the use of the GABRG3 Antibody (N-term) for immunohistochemistry. Storage Clinical relevance has not been evaluated. Maintain refrigerated at 2-8°C for up to 2 weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw cycles. Precautions Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 GABRG3 Antibody (N-term) is for research use only and not for use in diagnostic or therapeutic procedures. -

Kenneth Martin Rosenberg Email: [email protected], [email protected] 660 West Redwood Street, Howard Hall Room 332D, Baltimore, MD, 21201
The impact of the non-immune chemiome on T cell activation Item Type dissertation Authors Rosenberg, Kenneth Publication Date 2020 Abstract T cells are critical organizers of the immune response and rigid control over their activation is necessary for balancing host defense and immunopathology. It takes 3 signals provided by dendritic cells (DC) to fully activate a T cell response – T ce... Keywords signaling; T cell; T-Lymphocytes--immunology Download date 02/10/2021 13:41:58 Link to Item http://hdl.handle.net/10713/14477 Kenneth Martin Rosenberg Email: [email protected], [email protected] 660 West Redwood Street, Howard Hall Room 332D, Baltimore, MD, 21201 EDUCATION MD, University of Maryland, Baltimore, MD Expected May 2022 PhD, University of Maryland, Baltimore, MD December 2020 Graduate Program: Molecular Microbiology and Immunology (MMI) BS, University of Maryland, College Park, MD May 2013 Major: Bioengineering, cum laude University Honors Citation, Gemstone Citation RESEARCH EXPERIENCE UMSOM Microbiology and Immunology Baltimore, MD July 2016-present PhD Candidate Principal Investigator: Dr. Nevil Singh Thesis: The impact of the non-immune chemiome on T cell activation Examined environmental stimuli from classically “non-immune” sources – growth factors, hormones, neurotransmitters, etc. – act to modulate T cell signaling pathways and the functional effects of activating encounters with dendritic cells. UMSOM Anatomy and Neurobiology Baltimore, MD May-August 2015 Rotating student Principal Investigator: Dr. Asaf Keller Studied the role of descending modulation pathways on affective pain transmission. Performed tract- tracing experiments using targeted injection of Cholera toxin subunit B into the lateral parabrachial nucleus and ventrolateral periaqueductal gray of anesthetized transgenic mice. -

1 1 2 3 Cell Type-Specific Transcriptomics of Hypothalamic
1 2 3 4 Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to 5 weight-loss 6 7 Fredrick E. Henry1,†, Ken Sugino1,†, Adam Tozer2, Tiago Branco2, Scott M. Sternson1,* 8 9 1Janelia Research Campus, Howard Hughes Medical Institute, 19700 Helix Drive, Ashburn, VA 10 20147, USA. 11 2Division of Neurobiology, Medical Research Council Laboratory of Molecular Biology, 12 Cambridge CB2 0QH, UK 13 14 †Co-first author 15 *Correspondence to: [email protected] 16 Phone: 571-209-4103 17 18 Authors have no competing interests 19 1 20 Abstract 21 Molecular and cellular processes in neurons are critical for sensing and responding to energy 22 deficit states, such as during weight-loss. AGRP neurons are a key hypothalamic population 23 that is activated during energy deficit and increases appetite and weight-gain. Cell type-specific 24 transcriptomics can be used to identify pathways that counteract weight-loss, and here we 25 report high-quality gene expression profiles of AGRP neurons from well-fed and food-deprived 26 young adult mice. For comparison, we also analyzed POMC neurons, an intermingled 27 population that suppresses appetite and body weight. We find that AGRP neurons are 28 considerably more sensitive to energy deficit than POMC neurons. Furthermore, we identify cell 29 type-specific pathways involving endoplasmic reticulum-stress, circadian signaling, ion 30 channels, neuropeptides, and receptors. Combined with methods to validate and manipulate 31 these pathways, this resource greatly expands molecular insight into neuronal regulation of 32 body weight, and may be useful for devising therapeutic strategies for obesity and eating 33 disorders. -

An Observational and Genome-Wide by Environment Interaction Study in UK Biobank
Translational Psychiatry www.nature.com/tp ARTICLE OPEN Traumatic events during childhood and its risks to substance use in adulthood: an observational and genome-wide by environment interaction study in UK Biobank 1,2 1,2 1 1 1 1 1 1 1 Shiqiang Cheng , Yan Wen , Li Liu✉ , Bolun Cheng , Chujun Liang , Jing Ye , Xiaomeng Chu , Yao Yao , Yumeng Jia , Om Prakash Kafle1 and Feng Zhang 1 © The Author(s) 2021 We aimed to explore the underlying genetic mechanisms of traumatic events during childhood affecting the risks of adult substance use in present study. Using UK Biobank cohort, linear regression model was first applied to assess the relationships between cigarette smoking and alcohol drinking in adults with traumatic events during childhood, including felt hated by family member (41,648–111,465), felt loved (46,394–124,481) and sexually molested (47,598–127,766). Using traumatic events as exposure variables, genome-wide by environment interaction study was then performed by PLINK 2.0 to identify cigarette smoking and alcohol drinking associated genes interacting with traumatic events during childhood. We found that the frequency of cigarette smoking was significantly associated with felt hated by family member (coefficient = 0.42, P < 1.0 × 10–9), felt loved (coefficient = −0.31, P < 1.0 × 10–9) and sexually molested (coefficient = 0.46, P < 1.0 × 10–9). We also observed weaker associations of alcohol drinking with felt hated by family member (coefficient = 0.08, P = 3.10 × 10–6) and felt loved (coefficient = −0.06, P = 3.15 × 10–7). GWEIS identified multiple candidate loci interacting with traumatic events, such as CTNNA3 (rs189142060, P = 4.23 × 10–8) between felt hated by family member and the frequency of cigarette smoking, GABRG3 (rs117020886, P = 2.77 × 10–8) between felt hated by family member and the frequency of alcohol drinking.