FLIM) to Study Metabolism and Oxidative Stress in Biological Systems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rabindranath Tagore: a Social Thinker and an Activist a Review of Literature and a Bibliography Kumkum Chattopadhyay, Retd
2018 Heritage Vol.-V Rabindranath Tagore: a Social Thinker and an Activist A Review of Literature and a Bibliography Kumkum Chattopadhyay, Retd. Associate Professor, Dept. of Political Science, Bethune College, Kolkata-6 Abstract: Rabindranath Tagore, although basically a poet, had a multifaceted personality. Among his various activities his sincerity as a social thinker and activist attract our attention. But this area is till now comparatively unexplored. Many scholars in this area have tried to study Tagore as a social thinker. But so far the findings are scattered and on the whole there is no comprehensive analysis in the strict sense of the term. Hence it is necessary to collect the different findings and to integrate and arrange them within a theoretical framework. This article is an attempt to make a review of literature of the existing books and to prepare a short but sharp bibliography to introduce the area. Key words: Rabindranath Tagore, society, social, political, history, education, Santiniketan, Visva-Bharati Rabindranath Tagore (1861 – 1941) was a prolific writer, a successful music composer, a painter, an actor, a drama director and what not. Besides these talents, he was also a social activist and contributed a lot to Indian social and political thought, although this area has not been very much explored till now. Tagore was emphatic upon society building. So he tried to develop all the component elements which were essential for developing the Indian society. He studied the history of India to follow the trend of its evolution. Next he prepared his programme of action – rural reconstruction and spread of education. -

Nandan Gupta. `Prak-Bibar` Parbe Samaresh Basu. Nimai Bandyopadhyay
BOOK DESCRIPTION AUTHOR " Contemporary India ". Nandan Gupta. `Prak-Bibar` Parbe Samaresh Basu. Nimai Bandyopadhyay. 100 Great Lives. John Cannong. 100 Most important Indians Today. Sterling Special. 100 Most Important Indians Today. Sterling Special. 1787 The Grand Convention. Clinton Rossiter. 1952 Act of Provident Fund as Amended on 16th November 1995. Government of India. 1993 Vienna Declaration and Programme of Action. Indian Institute of Human Rights. 19e May ebong Assame Bangaliar Ostiter Sonkot. Bijit kumar Bhattacharjee. 19-er Basha Sohidera. Dilip kanti Laskar. 20 Tales From Shakespeare. Charles & Mary Lamb. 25 ways to Motivate People. Steve Chandler and Scott Richardson. 42-er Bharat Chara Andolane Srihatta-Cacharer abodan. Debashish Roy. 71 Judhe Pakisthan, Bharat O Bangaladesh. Deb Dullal Bangopadhyay. A Book of Education for Beginners. Bhatia and Bhatia. A River Sutra. Gita Mehta. A study of the philosophy of vivekananda. Tapash Shankar Dutta. A advaita concept of falsity-a critical study. Nirod Baron Chakravarty. A B C of Human Rights. Indian Institute of Human Rights. A Basic Grammar Of Moden Hindi. ----- A Book of English Essays. W E Williams. A Book of English Prose and Poetry. Macmillan India Ltd.. A book of English prose and poetry. Dutta & Bhattacharjee. A brief introduction to psychology. Clifford T Morgan. A bureaucrat`s diary. Prakash Krishen. A century of government and politics in North East India. V V Rao and Niru Hazarika. A Companion To Ethics. Peter Singer. A Companion to Indian Fiction in E nglish. Pier Paolo Piciucco. A Comparative Approach to American History. C Vann Woodward. A comparative study of Religion : A sufi and a Sanatani ( Ramakrishana). -

Dishti the Sign 2019 Final.Pmd
Drishti: the Sight Vol.VIII, Issues: I & II(Combined volume) (May, 2019 - October, 2019) & (November, 2019 - April, 2020) ISSN 2319-8281 (Enlisted in the UGC-CARE list (Sl. No. 42) in Arts and Humanities section) A REFEREED (PEER-REVIEWED) BI-ANNUAL NATIONAL RESEARCH JOURNAL OF ENGLISH LITERATURE/ASSAMESE LITERATURE/FOLKLORE /CULTURE Chief Editor (Hon.) DR. DIPAK JYOTI BARUAH Associate Professor, Dept. of English, Jagiroad College(University of Gauhati) Associate Editors : Dr. Manash Pratim Borah (Dept. of English, Central Institute of Himalayan Culture Studies, Arunachal Pradesh) Dr. Nizara Hazarika (Dept. of English, Sonapur College, University of Gauhati) Dr. Bhubaneswar Deka (Dept. of English, Pandu College, University of Gauhati) Members of Advisory Body Dr. Gayatree Bhattacharyya, Former Professor, Dept. of English, University of Gauhati Dr. Dayananda Pathak, Former Principal, Pragjyotish College, Guwahati Dr. Prabin Chandra Das, Former Head, Dept. of Folklore, University of Gauhati Dr. Dwijen Sharma, Professor, Dept. of English, North Eastern Hill University (Tura Campus) Dr. Kalikinkar Pattanayak, Former Associate Professor in English, Khallikote University, Odisha Members of Editorial Body Dr. Pranjal Sharma Bashistha, Dept. of Assamese, University of Gauhati Mr. Lakshminath Kagyung, Dept. of English, University of Dibrugarh Dr. Lakshmi Dash Pathak, Dept. of Assamese, L.C.B College, University of Gauhati Mr. Pranjal Dutta, Dept. of English, Sarupathar College, University of Dibrugarh Dr. Durga Prasad Dash, Lecturer, Dept. of English, Gunjam College, Berhapur University, Odisha Address for correspondence: Dr. Dipak Jyoti Baruah, Associate Professor, Dept.of English, Jagiroad College, Jagiroad-782410, Assam, India; Cell: 09854369647; E-mail: [email protected]/[email protected] Our website : www.drishtithesight.com Drishti: the Sight Vol.VIII, Issues: I & II(Combined volume) (May, 2019 - October, 2019) & (November, 2019 - April, 2020) ISSN 2319-8281 (Enlisted in the UGC-CARE list (Sl. -
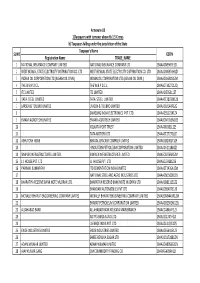
FINAL DISTRIBUTION.Xlsx
Annexure-1B 1)Taxpayers with turnover above Rs 1.5 Crores b) Taxpayers falling under the jurisdiction of the State Taxpayer's Name SL NO GSTIN Registration Name TRADE_NAME 1 NATIONAL INSURANCE COMPANY LIMITED NATIONAL INSURANCE COMPANY LTD 19AAACN9967E1Z0 2 WEST BENGAL STATE ELECTRICITY DISTRIBUTION CO. LTD WEST BENGAL STATE ELECTRICITY DISTRIBUTION CO. LTD 19AAACW6953H1ZX 3 INDIAN OIL CORPORATION LTD.(ASSAM OIL DIVN.) INDIAN OIL CORPORATION LTD.(ASSAM OIL DIVN.) 19AAACI1681G1ZM 4 THE W.B.P.D.C.L. THE W.B.P.D.C.L. 19AABCT3027C1ZQ 5 ITC LIMITED ITC LIMITED 19AAACI5950L1Z7 6 TATA STEEL LIMITED TATA STEEL LIMITED 19AAACT2803M1Z8 7 LARSEN & TOUBRO LIMITED LARSEN & TOUBRO LIMITED 19AAACL0140P1ZG 8 SAMSUNG INDIA ELECTRONICS PVT. LTD. 19AAACS5123K1ZA 9 EMAMI AGROTECH LIMITED EMAMI AGROTECH LIMITED 19AABCN7953M1ZS 10 KOLKATA PORT TRUST 19AAAJK0361L1Z3 11 TATA MOTORS LTD 19AAACT2727Q1ZT 12 ASHUTOSH BOSE BENGAL CRACKER COMPLEX LIMITED 19AAGCB2001F1Z9 13 HINDUSTAN PETROLEUM CORPORATION LIMITED. 19AAACH1118B1Z9 14 SIMPLEX INFRASTRUCTURES LIMITED. SIMPLEX INFRASTRUCTURES LIMITED. 19AAECS0765R1ZM 15 J.J. HOUSE PVT. LTD J.J. HOUSE PVT. LTD 19AABCJ5928J2Z6 16 PARIMAL KUMAR RAY ITD CEMENTATION INDIA LIMITED 19AAACT1426A1ZW 17 NATIONAL STEEL AND AGRO INDUSTRIES LTD 19AAACN1500B1Z9 18 BHARATIYA RESERVE BANK NOTE MUDRAN LTD. BHARATIYA RESERVE BANK NOTE MUDRAN LTD. 19AAACB8111E1Z2 19 BHANDARI AUTOMOBILES PVT LTD 19AABCB5407E1Z0 20 MCNALLY BHARAT ENGGINEERING COMPANY LIMITED MCNALLY BHARAT ENGGINEERING COMPANY LIMITED 19AABCM9443R1ZM 21 BHARAT PETROLEUM CORPORATION LIMITED 19AAACB2902M1ZQ 22 ALLAHABAD BANK ALLAHABAD BANK KOLKATA MAIN BRANCH 19AACCA8464F1ZJ 23 ADITYA BIRLA NUVO LTD. 19AAACI1747H1ZL 24 LAFARGE INDIA PVT. LTD. 19AAACL4159L1Z5 25 EXIDE INDUSTRIES LIMITED EXIDE INDUSTRIES LIMITED 19AAACE6641E1ZS 26 SHREE RENUKA SUGAR LTD. 19AADCS1728B1ZN 27 ADANI WILMAR LIMITED ADANI WILMAR LIMITED 19AABCA8056G1ZM 28 AJAY KUMAR GARG OM COMMODITY TRADING CO. -

The Pen Souvenir Revolt
THE PEN SOUVENIR REVOLT underground literature published during the emergency A Letter from a Father to his Son written by Gour Kishore Ghosh, from prison The author of the following letter is a journalist of repute on the staff of Calcutta's "Ananda Bazar Patrika". He was arrested on October 6, 1975 for allowing this letter to be published in Kolkata, a Bengali monthly edited by Jyotirmoy Datta. Later Bhumiputra of Baroda was asked to deposit a security of Rs. 25,000 for publishing the Gujarati version of this letter. Copies of it were made and circulated by the underground network. So you understand that I resent very much the fact dean that my freedom as a writer has been taken away; I For some time now I have been thinking of having a cannot reconcile myself to this situation. Not merely few words with you about myself. For about two that, I want to register publicly my protest against this months now something has been troubling me. I find wrong. But whenever I think of protesting, your face, no remedy for it. The only relief that I can expect is to the faces of your sisters, of your mother and of your unburden my mind to you. grand-mother all appear before my minds' eye. Some of At your age —you are only thirteen—much of what I you are young. Some helpless and some old. All of you shall speak now may seen incomprehensible. Yet the are dependent on me. All these years I have tried to reason I write to you alone of my trouble is that you provide you with a secure home. -

Sunil Gangopadhyay Wikipedia Sunil Gangopadhyay from Wikipedia, the Free Encyclopedia
6/8/2017 Sunil Gangopadhyay Wikipedia Sunil Gangopadhyay From Wikipedia, the free encyclopedia Sunil Gangopadhyay or Sunil Ganguly (Bengali: সুনীল গেЀাপাধᘃায় Shunil Gônggopaddhae), (7 September 1934 – 23 Sunil Gangopadhyay October 2012)[2] was an Indian Bengali poet and novelist based in the Indian city of Kolkata.[4] Born in Faridpur, in what is now Bangladesh, Gangopadhyay obtained his master's degree in Bengali from the University of Calcutta. In 1953 he and a few of his friends started a Bengali poetry magazine, Krittibas. Later he wrote for many different publications. Ganguly created the Bengali fictional character Kakababu and wrote a series of novels on this character which became significant in Indian children's literature. He received Sahitya Akademi award in 1985 for his novel Those Days (Sei Samaya).[5] Gangopadhyay used the pen names Nil Lohit, Sanatan Pathak, and Nil Upadhyay.[2] Contents Born 7 September 1934 Faridpur, Bengal Presidency, British 1 Early life India (Now Bangladesh)[1] 2 Literary career Died 23 October 2012 (aged 78) 2.1 Krittibas Kolkata, India 2.2 Other works 3 Works Pen name Nil Lohit, Sanatan Pathak, and Nil 3.1 Film based on his literary works Upadhyay[2] 4 Death Occupation Writer 5 Controversies 6 List of major works Language Bengali 6.1 Poetry Nationality Indian 6.2 Novels Ethnicity Bengali 6.3 Autobiography 6.3.1 Kakababu series Education Master of Arts (Bengali) 6.4 Translated books Alma mater University of Calcutta (1954) 7 Awards and honours Period 1953–2012 7.1 Awards 7.2 Honors Notable First Light (Prathama Alo), Those 8 See also works Days (Sei Somoy), East and West 9 Notes (Poorba Pashchim) 10 External links Notable Ananda Puraskar (1972, 1989) awards Sahitya Akademi Award (1985) Spouse Swati Bandopadhyay (m. -

The High Court at Calcutta 150 Years : an Overview
1 2 The High Court at Calcutta 150 Years : An Overview 3 Published by : The Indian Law Institute (West Bengal State Unit) iliwbsu.in Printed by : Ashutosh Lithographic Co. 13, Chidam Mudi Lane Kolkata 700 006 ebook published by : Indic House Pvt. Ltd. 1B, Raja Kalikrishna Lane Kolkata 700 005 www.indichouse.com Special Thanks are due to the Hon'ble Justice Indira Banerjee, Treasurer, Indian Law Institute (WBSU); Mr. Dipak Deb, Barrister-at-Law & Sr. Advocate, Director, ILI (WBSU); Capt. Pallav Banerjee, Advocate, Secretary, ILI (WBSU); and Mr. Pradip Kumar Ghosh, Advocate, without whose supportive and stimulating guidance the ebook would not have been possible. Indira Banerjee J. Dipak Deb Pallav Banerjee Pradip Kumar Ghosh 4 The High Court at Calcutta 150 Years: An Overview तदॆततत- क्षत्रस्थ क्षत्रैयद क्षत्र यद्धर्म: ।`& 1B: । 1Bद्धर्म:1Bत्पटैनास्ति।`抜֘टै`抜֘$100 नास्ति ।`抜֘$100000000स्ति`抜֘$1000000000000स्थक्षत्रैयदत । तस्थ क्षत्रै यदर्म:।`& 1Bण । ᄡC:\Users\सत धर्म:" ।`&ﲧ1Bशैसतेधर्मेण।h अय अभलीयान् भलीयौसमाशयनास्ति।`抜֘$100000000 भलीयान् भलीयौसमाशयसर्म: ।`& य राज्ञाज्ञा एवम एवर्म: ।`& 1B ।। Law is the King of Kings, far more powerful and rigid than they; nothing can be mightier than Law, by whose aid, as by that of the highest monarch, even the weak may prevail over the strong. Brihadaranyakopanishad 1-4.14 5 Copyright © 2012 All rights reserved by the individual authors of the works. All rights in the compilation with the Members of the Editorial Board. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording or any information storage and retrieval system, without permission from the copyright holders. -

Six Korean Poets in Translation
Shabdaguchha, Issue 51/52 An International Bilingual Poetry Journal Vol. 13 No. 3/4 January–June 2011 $5 Editor: Hassanal Abdullah January - June 2011 $5 Advisor: Jyotirmoy Datta www.shabdaguchha.com Assistant Editor: Naznin Seamon Consulting Editor: Leigh Harrison Correspondents: AKM Mizanur Rahman (Bangladesh) Prabir Das (India) Nazrul Islam Naz (London) Special Correspondent: Romel Rahman (Dhaka) Copyright © 2011 by Shabdaguchha Press All right Reserved Shabdaguchha, a Bilingual (Bengali-English) Poetry Quarterly, Published by the editor, from Shabdaguchha Press. Subscription rate is $12 per year in the United States and $15 in other countries. To subscribe, please send check or money order in US Dollar payable to Shabdaguchha. All correspondence and subscriptions should be addressed to the editor: Shabdaguchha, 85-22 85th street, Woodhaven, NY 11421, USA. Phone: (718) 849 2617 E-mail: [email protected] [email protected] Web Site: http://www.shabdaguchha.com Six Korean Poets Shabdaguchha accepts submission throughout the whole year. Poetry, written in Bengali, English or translated from any language to these two languages, is always welcome. Book review and news on in poets and poetry could also be sent. Each submission should accompany with a short bio of the author. E-mail submissions are more appreciated, but Bengali written in English alphabet is not Issue 51/52 acceptable. Translation Shabdaguchha Press Woodhaven, New York ISSN 1531-2038 Editor: Hassanal Abdullah Cover Art: Jyotirmoy Datta 2 Shabdaguchha , an International Poetry Journal in Bengali and English Shabdaguchha Shabdaguchha, Issue 51/52 Editorial: Yoon-Ho Cho GB msL¨vwU cÖKv‡ki gva¨‡g k㸔Q i † Z‡iv eQi c~Y© n‡jv| GK BLUE POND hy‡MiI †ekx mgq a‡i my¯’ fv‡e cwÎKvwU cÖKvk Ki‡Z †c‡i Avgiv Under the sultry summer sun, Avbw›`Z| GB c_ Pjvq bexb cÖexY A‡bK KweB Avgv‡`i mv‡_ hy³ n‡q‡Qb, Avevi †KD †KD †Q‡oI †M‡Qb| m¤úK© wewbg©v‡Yi †¶‡Î the water in the pond mvaviYZ b`xi DÌvb cZ‡bi Zyjbv Kiv n‡jI, gbyl¨ †hvMv‡hvM mg‡qi slowly evaporates. -

Annual Report 2014-2015
VISVA-BHARATI Annual Report 2014-2015 Santiniketan 2015 YATRA VISVAM BHAVATYEKANIDAM (Where the World makes its home in a single nest) “ Visva-Bharati represents India where she has her wealth of mind which is for all. Visva-Bharati acknowledges India's obligation to offer to others the hospitality of her best culture and India's right to accept from others their best ” -Rabindranath Tagore Contents Chapter I .............................................................i-v Department of Chemistry .......................................60 From Bharmacharyashrama to Visva-Bharati.............i Department of Mathematics ...................................64 Institutional Structure Today ...................................ii Department of Zoology ..........................................69 Socially Relevant Research and Other Activities ......iii Department of Botany ............................................73 Finance ................................................................... v Department of Statistics .........................................77 Administrative Staff Composition .............................v Department of Computer & System Sciences...........79 University At a Glance................................................v Department of Environmental Studies ...................81 Student Composition .................................................v Department of Biotechnology ...............................84 Teaching Staff Composition ......................................v Centre for Mathematics Education .........................87 -

Rhixltspreads.Pdf
UNCOVERAGE: asking after recent poetry interviews by H. L. HIX #45 CONTENTS ESSAY PRESS EP SERIES In the Essay Press EP Series, we give extended In Memoriam ix space and time to some of our favorite authors currently developing new book-length projects. Introduction xi Interviews Ali Alizadeh on Eyes in Times of War 1 Series Editors Maria Anderson Dan Beachy-Quick on This Nest, Swift Passerine 6 Andy Fitch Sherwin Bitsui on Shapeshift 11 Ellen Fogelman Aimee Harrison Rebecca Black on Cottonlandia 14 Courtney Mandryk Shane Book on Ceiling of Sticks 19 Victoria A. Sanz Jericho Bown on Please 29 Travis A. Sharp Julie Carr on 100 Notes on Violence 33 Ryan Spooner Jennifer Chang on The History of Anonymity 39 Randall Tyrone Justin Chin on Gutted 43 Don Mee Choi on Kim Hyesoon’s Series Assistants Cristiana Baik Mommy Must Be a Fountain of Feathers 47 Ryan Ikeda Allison Adelle Hedge Coke on Effigies: An Anthology of Christopher Liek New Indigenous Writing, Pacific Rim, 2009 53 Mary Dalton on Merrybegot 56 Cover Design Courtney Mandryk Sesshu Foster on World Ball Notebook 63 Layout Aimee Harrison Santee Frazier on Dark Thirty 65 Asher Ghaffar on Wasps in a Golden Dream Hum a Strange Music 69 Ferial Ghazoul on Muhammad Afifi Matar’s Quartet of Joy 73 Johannes Göransson on Aase Berg’s Remainland 77 Noah Eli Gordon on A Fiddle Pulled from the Throat of a Sparrow 82 Kimiko Hahn on The Narrow Road to the Interior 87 Jesse Nathan on Dinner 201 Sam Hamill on Avocations 90 Uche Nduka on eel on reef 207 Robert Hass on Time and Materials 94 Maggie Nelson -

S. J. Chemicals Private Limited
+91-8048424322 S. J. Chemicals Private Limited https://www.indiamart.com/sjchemicals/ Established in the year of 1987, S. J. Chemicals Pvt. Ltd. is the leading wholesale trader and exporter of Chemical Paste, Chemical Liquid, Chemical Flakes and much more. About Us Established in the year of 1987, S. J. Chemicals Pvt. Ltd. is the leading wholesale trader and exporter of Chemical Paste, Chemical Liquid, Chemical Flakes and much more. These are extremely admired in the market due to their top features. These products are widely admired among our esteemed patrons owing to their eco-friendly, excellent quality and various packing options. These products are made in adherence with worldwide quality principles using the best quality of chemical input and advanced techniques. Beside this, clientele can easily buy these products from us at nominal prices. We work under the guidance of our mentor Mr. Jyotirmoy Datta. Under his guidance, we have gained fame and glory in this area. For more information, please visit https://www.indiamart.com/sjchemicals/profile.html CHEMICAL LIQUID O u r P r o d u c t R a n g e Sodium Lauryl Sulphate Liquid Monoethylene Glycol Oleic Acid Sodium Lauryl Ether Sulphate CHEMICAL POWDER O u r P r o d u c t R a n g e Sodium Lauryl Sulphate Acid Thickener Powder Cocamidopropyl Betaine CHEMICAL FLAKES O u r P r o d u c t R a n g e Ethylene Glycol Monostearate Emulsifying Wax Cationic Softener O u r OTHER PRODUCTS: P r o d u c t R a n g e Benzalkonium Chloride Sodium Lauryl Sulphate Paste Solution 50% Hand Sanitizer Butyl Carbitol F a c t s h e e t Year of Establishment : 1987 Nature of Business : Wholesale Trader Total Number of Employees : Upto 10 People CONTACT US S. -

Sunil Gangopadhyay - Poems
Classic Poetry Series Sunil Gangopadhyay - poems - Publication Date: 2012 Publisher: Poemhunter.com - The World's Poetry Archive Sunil Gangopadhyay(7 September 1934 -) Sunil Gangopadhyay (Shunil Gônggopaddhae), is an Indian poet and novelist. <b>Early Life</b> He was born in Faridpur in what is now Bangladesh. He studied at the Surendranath College, Dumdum Motizhil College, City College, Kolkata - all affiliated with the University of Calcutta. Thereafter, he obtained his Master's degree in Bengali from the University of Calcutta in 1954. <b>Literary Career</b> He was the founder editor of Krittibas, a seminal poetry magazine started publishing from 1953, that became a platform for a new generation of poets experimenting with many new forms in poetic themes, rhythms, and words. Later, he started writing for various publications of the Ananda Bazar group, a major publishing house in Kolkata and has been continuing it for many years. He became friends with the beat poet Allen Ginsberg while he was traveling in India. Ginsberg mentioned Gangopadhyay most notedly in his poem September on Jessore Road. Gangopadhyay in return mentioned Ginsberg in some of his prose work. After serving five years as the Vice President, he was elected the President of the Sahitya Akademi on February 20, 2008. Sunil, along with Tarun Sanyal, Jyotirmoy Datta and Satrajit Dutta had volunteered to be defense witnesses in the famous trial of "Hungryalism" poet Malay Roy Choudhury. <b>Personal Life</b> He married Swati Bandopadhyay on February 26, 1967. Their only son, Sauvik was born on November 20, 1967. <b>Works</b> Author of well over 200 books, Sunil is a prolific writer who has excelled in different genres but declares poetry to be his "first love".