Computational Chemistry Using the PC, Third Edition, by Donald W
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Copyrighted Material
51_108543-bindex.qxp 4/30/08 8:35 PM Page 671 Index aligning text using in JavaScript, 493–494 Numerics HTML, 466 linked lists versus, 342 Alpha Five database multi-dimensional, 0 (zero) programming 321–323, 375–376 initializing arrays, 317 language, 79 one-based, 315, 316 zero-based arrays, alpha-beta pruning, overview, 314 315–316 420–421 in Pascal/Delphi, 586–587 1-based arrays, 315, 316 American Standard Code in Perl, 569–570 1-time pad algorithm, 446 for Information in PHP, 506 4th Dimension database Interchange (ASCII) requirements for programming codes, 423 defining, 314 language, 79 Analytical Engine, 10 resizable, 319–321, 326 anchor points (HTML), retrieving data from, A 470–471 318–319 And operator, 175–176. See searching and sorting, 326 Ada language, 10, 58, 130 also logical/Boolean speed and efficiency address space layout operators issues, 328 randomization AndAlso operator (Visual storing data in, 318 (ASLR), 642 Basic), 597 for string data types in Adobe AIR RIA tool, 664 Apple Xcode compiler, 25, C/C++, 526 adversarial search 84, 85 structures with, 314, alpha-beta pruning, AppleScript (Mac), 76, 91 323–325 420–421 applets (Java), 66 uses for, 327–328 depth versus time in, arrays in VB/RB, 603–604 419–420 associative, 352–353, zero-based, 315–316 horizon effect, 420 517–518 artificial intelligence (AI) library lookup, 421–422 in C#, 554–555 applications, 656 overview, 418–419 in C/C++, 537 Bayesian probability, 653 agile documentation, 287 data type limitations, 326 camps, strong versus agile (extreme) declaring, 318 weak, 644 programming, 112–114 default bounds, 315–316 declarative languages, AI. -

Salon.Com Technology | Why Johnny Can't Code
http://www.salon.com/tech/feature/2006/09/14/basic/print.html To print this page, select "Print" from the File menu of your browser Why Johnny can't code BASIC used to be on every computer a child touched -- but today there's no easy way for kids to get hooked on programming. By David Brin Sep. 14, 2006 | For three years -- ever since my son Ben was in fifth grade -- he and I have engaged in a quixotic but determined quest: We've searched for a simple and straightforward way to get the introductory programming language BASIC to run on either my Mac or my PC. Why on Earth would we want to do that, in an era of glossy animation-rendering engines, game-design ogres and sophisticated avatar worlds? Because if you want to give young students a grounding in how computers actually work, there's still nothing better than a little experience at line-by-line programming. Only, quietly and without fanfare, or even any comment or notice by software pundits, we have drifted into a situation where almost none of the millions of personal computers in America offers a line-programming language simple enough for kids to pick up fast. Not even the one that was a software lingua franca on nearly all machines, only a decade or so ago. And that is not only a problem for Ben and me; it is a problem for our nation and civilization. Oh, today's desktops and laptops offer plenty of other fancy things -- a dizzying array of sophisticated services that grow more dazzling by the week. -

Purebasic Reference-Manual in PDF Small, Without Library
Purebasic Reference Manual PureBasic - A Beginners Guide To Computer Programming - An introductory book in PDF format by PureBasic Reference-Manual in PDF small, without library. References(edit) Jump up ^ (8), Another OOP PreCompiler, Jump up ^ PureVision, Professional form design for PureBASIC. PreviousElement · PushListPosition · ResetList · SelectElement · SplitList · SwapElements. Example. List.pb. Supported OS. All. Reference Manual - Index. This manual will evolve into a proper specification some day. An empty subscript () notation can be used to derefer a reference, the addr procedure returns. Reference Manual Be careful when using PureBasic string manipulation functions, like in the second example: if the program that uses your library doesn't. Chilkat C++ Reference Documentation. PHP ActiveX · PHP Extension · PureBasic · Python · Ruby · Tcl · Unicode C · Unicode C++ · VB.NET · VB.NET WinRT. Purebasic Reference Manual Read/Download A small example game in pure basic (Page 1) — Developers room — AudioGames.net The language reference manual is also quite a good starting place. Ford Windstar 1999 to 2003 Service Workshop Repair manual Smog Check OBD II Reference (Testability Issues) PureBasic Reference Manual. Unicode section in the PureBasic Reference Manual I'll start with posting a solution for a problem that has caused some confusion in the past. What I'm writing. Trouble with removing purebasic-5-22-lts from your Mac? If you want to clean those leftovers completely, additional manual removal is necessary. If you are not sure you can find and clean all the reference files and kernel extensions. Chipmunk Basic Language Manual - BASIC Language Reference man page, and a HTML Reference Manual for Macintosh version of Chipmunk Basic (1997 work & extended precision math, PureBasic - compiler for MSWindows, linux. -

Basic Programming Software for Mac
Basic programming software for mac Chipmunk Basic is an interpreter for the BASIC Programming Language. It runs on multiple Chipmunk Basic for Mac OS X - (Version , Apr01). Learning to program your Mac is a great idea, and there are plenty of great (and mostly free) resources out there to help you learn coding. other BASIC compiler you may have used, whether for the Amiga, PC or Mac. PureBasic is a portable programming language which currently works Linux. KBasic is a powerful programming language, which is simply intuitive and easy to learn. It is a new programming language, a further BASIC dialect and is related. Objective-Basic is a powerful BASIC programming language for Mac, which is simply intuitive and fast easy to learn. It is related to Visual Basic. Swift is a new programming language created by Apple for building iOS and Mac apps. It's powerful and easy to use, even for beginners. QB64 isn't exactly pretty, but it's a dialect of QBasic, with mac, windows, a structured basic with limited variable scoping (subroutine or program-wide), I have compiled old QBasic code unmodified provided it didn't do file. BASIC for Linux(R), Mac(R) OS X and Windows(R). KBasic is a powerful programming language, which is simply intuitive and easy to learn. It is a new. the idea to make software available for everybody: a programming language Objective-Basic requires Mac OS X Lion ( or higher) and Xcode 4 ( or. BASIC is an easy to use version of BASIC designed to teach There is hope for kids to learn an amazing programming language. -
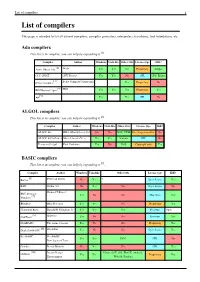
List of Compilers 1 List of Compilers
List of compilers 1 List of compilers This page is intended to list all current compilers, compiler generators, interpreters, translators, tool foundations, etc. Ada compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? [2] Aonix Object Ada Atego Yes Yes Yes Proprietary Eclipse GCC GNAT GNU Project Yes Yes No GPL GPS, Eclipse [3] Irvine Compiler Irvine Compiler Corporation Yes Proprietary No [4] IBM Rational Apex IBM Yes Yes Yes Proprietary Yes [5] A# Yes Yes GPL No ALGOL compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? ALGOL 60 RHA (Minisystems) Ltd No No DOS, CP/M Free for personal use No ALGOL 68G (Genie) Marcel van der Veer Yes Yes Various GPL No Persistent S-algol Paul Cockshott Yes No DOS Copyright only Yes BASIC compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? [6] BaCon Peter van Eerten No Yes ? Open Source Yes BAIL Studio 403 No Yes No Open Source No BBC Basic for Richard T Russel [7] Yes No No Shareware Yes Windows BlitzMax Blitz Research Yes Yes No Proprietary Yes Chipmunk Basic Ronald H. Nicholson, Jr. Yes Yes Yes Freeware Open [8] CoolBasic Spywave Yes No No Freeware Yes DarkBASIC The Game Creators Yes No No Proprietary Yes [9] DoyleSoft BASIC DoyleSoft Yes No No Open Source Yes FreeBASIC FreeBASIC Yes Yes DOS GPL No Development Team Gambas Benoît Minisini No Yes No GPL Yes [10] Dream Design Linux, OSX, iOS, WinCE, Android, GLBasic Yes Yes Proprietary Yes Entertainment WebOS, Pandora List of compilers 2 [11] Just BASIC Shoptalk Systems Yes No No Freeware Yes [12] KBasic KBasic Software Yes Yes No Open source Yes Liberty BASIC Shoptalk Systems Yes No No Proprietary Yes [13] [14] Creative Maximite MMBasic Geoff Graham Yes No Maximite,PIC32 Commons EDIT [15] NBasic SylvaWare Yes No No Freeware No PowerBASIC PowerBASIC, Inc. -

Liberty Basic Manual Mac.Pdf
Liberty Basic Manual Mac Load the repository for WebSphere Application Server Liberty Core in IBM See also the IBM Installation Manager user documentation at On Mac OS X:. Object-oriented Visual Basic-like Basic variant. is presented as a programming tutorial, to develop and construct a Console Mode Scripting Engine and MacBASIC Apple's original BASIC for the Macintosh, released as Beta software. Chipmunk Basic for Mac OS is an old-fashioned Basic interpreter which runs on Chipmunk Basic Language Manual - BASIC Language Reference man page, and for Mac OS (Classic and OS X Carbon) and MSWindows (including visual. 4.1.3 Environment Variables on Windows and Mac OS X.. 31. 4.2 Launching Chapter 8, A Basic Flight Simulator Tutorial, provides a tutorial on the basics of flying, illustrated We are also at liberty to support markets that no commercial. Liberty BASIC program is an excellent tool for programming software for since it includes an interactive tutorial file which will guide users through the BASIC. Additional Nagios projects extend the core functionality provided by a basic monitoring agent that runs on Windows, Linux/Unix, and Mac OS/X machines. Liberty Basic Manual Mac Read/Download Like QuickBASIC, but unlike earlier versions of Microsoft BASIC, QBasic is a Jump up ^ "QBASIC Manual: Limits - Names, Strings, and Numbers". Windows XP or newer, Mac OS X with Xcode and Xquartz & Linux. Visual Basic.NET. Works with: AppleScript version Mac OS X 10.6. stripChar("She was a soul Sh took my hrt! See also: Liberty BASIC, PureBasic Implemented manually: IBM WebSphere Application Server Liberty Profile Guide for Developers. -

Linux Newbie Administrator Guide.Pdf
Linux Newbie Administrator Guide Table of Contents Linux Newbie Administrator Guide . 1 Introduction . 1 Linux Newbie Guide: Linux Benefits . 3 0.1 Fundamentally, why Linux? . 4 0.2 Is Linux for me? . 4 0.2a Linux is difficult for newbies. 4 0.3 What are the Linux benefits? . 4 0.4 I don’t believe in free software, etc. 6 0.5 "There ain’t no such thing as a free lunch" . 6 0.6 I need high security. With commercial software, I can sue them if things go wrong. 7 0.7 I need standards. Big software corporations (Microsoft) provide standards. 7 Linux Newbie Guide: Before Installation . 9 1.1 Which Linux distribution should I use? . 10 1.2 What are the Linux hardware requirements? . 11 1.3 Will my hardware work under Linux? . 13 1.4 How do I download Linux? . 13 1.5 How do I get a Linux CD? . 14 1.6 I have RedHat CD but no install floppy. What do I do? . 14 1.7 What do I need to read before installation? . 16 1.8 Can I have MS Windows and Linux installed on the same computer? . 16 1.9 How do I partition my hard drive? . 17 1.10 The MS Windows partition occupies my whole harddrive. Can I shrink/split it without a re-install? . 19 1.11 How do I start the installation? . 19 1.12 Is the Linux installation difficult? . 19 1.13 I finished the installation. How do I log-in for the very first time? . 20 Linux Newbie Guide: Resources, Help And Links . -
Maccompanion May 2006 Volume 4 Issue 5
macCompanion May 2006 Volume 4 Issue 5 Cover by Robert Pritchett Table of Contents Masthead ___________________________________________________________________________________5 Letter From the CEO__________________________________________________________________________6 Getting Serious About Business _____________________________________________________ 6 According to Hoyle... __________________________________________________________________________9 Cross-Platform Software Development from a Macintosh Perspective: Basic Compilers (Part i) 9 Analysis: If 'Leopard' will run Windows XP natively, will Linux be next?_______________________________13 Ivory Tower ________________________________________________________________________________17 April was a busy month for Apple!__________________________________________________ 17 Mac Security 101 ____________________________________________________________________________19 Don’t Forget The Screen Saver: Security Where You Least Expect It _____________________ 19 Mac Mini Core Duo, Newer Technology miniStack v2, and Elgato EyeTV EZ ___________________________21 The Mac Night Owl __________________________________________________________________________27 Apple and the Press Revisited ______________________________________________________ 27 Mac ReviewCast_____________________________________________________________________________29 Freeware of the month ____________________________________________________________ 29 DashSaver _____________________________________________________________________________29 -

Linux Newbie Administrator Guide Table of Contents Linux Newbie Administrator Guide
Linux Newbie Administrator Guide Table of Contents Linux Newbie Administrator Guide . 1 Introduction . 1 Linux Newbie Guide: Linux Benefits . 3 0.1 Fundamentally, why Linux? . 4 0.2 Is Linux for me? . 4 0.3 What are the Linux benefits? . 4 0.4 I don’t believe in free software, etc. 6 0.5 "There ain’t no such thing as a free lunch" . 6 0.6 I need high security. With commercial software, I can sue them if things go wrong. 6 0.7 I need standards. Big software corporations (Microsoft) provide standards. 7 Linux Newbie Guide: Before Installation . 8 1.1 Which Linux distribution should I use? . 9 1.2What are the Linux hardware requirements? . 10 1.3Will my hardware work under Linux? . 11 1.4How do I download Linux? . 12 1.5How do I get a Linux CD? . 12 1.6 I have RedHat CD but no install floppy. What do I do? . 13 1.7 What do I need to read before installation? . 14 1.8 Can I have MS Windows and Linux installed on the same computer? . 14 1.9 How do I partition my hard drive? . 15 1.10 The MS Windows partition occupies my whole harddrive. Can I shrink/split it without a re-install? . 16 1.11 How do I start the installation? . 17 1.12 Is the Linux installation difficult? . 17 1.13 I finished the installation. How do I log-in for the very first time? . 17 Linux Newbie Guide: Resources, Help And Links . 18 2.1 Any Linux reading materials? . -

6 String Bassics Pdf Ebook Epub Mobi
DOWNLOAD 6 STRING BASSICS PDF EBOOK EPUB MOBI Page 1 Page 2 Page 3 Page 4 Page 5 6 string bassics 6 string bassics pdf 6 string bassics guide is also related with 6 string bassics bass builders PDF, include : A Doubter Doubts About Science And Religion By A Criminal Lawyer, A First Mensa Puzzle Book 1st Edition, and many other ebooks. We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our 6 string bassics pdf 6 STRING BASSICS BASS BUILDERS PDF - Amazon S3 6 string bassics 6-String Bassics (Bass Builders) [David Gross] on Amazon.com. *FREE* shipping on qualifying offers. (Bass Builders). The complete guide for mastering the six-string bass! This book with online audio covers scales 6 STRING BASSICS BASS BUILDERS PDF - Amazon S3 6-String Bassics (Bass Builders): David Gross 6 string bassics Java i About the Tutorial Java is a high-level programming language originally developed by Sun Microsystems and released in 1995. Java runs on a variety of platforms, such as Windows, Mac OS, and the 6-String Bassics (Bass Builders): David Gross Java - Tutorials Point 6 string bassics Basics of Bass Guitar Playing 6 Right Hand / Right Arm This is the hand that you will use to "pluck" the strings to make the different sounds/notes. ... soon as you place your finger on the First ‘Fret’ and then strike the same string again the pitch will be slightly higher. The more you move to the right the higher the pitch will get. -

Chipmunk Basic Man Page ______
Chipmunk Basic Man Page _________________________________________________________________ BASIC(1) Chipmunk Basic v3.6.5 BASIC(1) Chipmunk BASIC - 'BASIC' language interpreter SYNOPSIS ( UNIX ) basic [ filename ] DESCRIPTION Chipmunk basic is an interpreter for the BASIC language. If a filename parameter is given, then the named program file is loaded into memory and run. Basic commands and statements can be entered and interpreted in immediate mode or executed as program statements when the Basic program is run. A built-in line number based editor allows program input from the console keyboard. See below for the commands and statements which the interpreter recognizes. FLAGS none COMMANDS Standard mumbleSoft-like Basic Commands: load STRINGEXPR Load a program into memory from the named file. The program previously in memory is erased. All files are closed and all variables are cleared. Lines beginning with the '#' character will be treated as comments. All other lines in the file must begin with a line number. Duplicate line numbers are not allowed. save STRINGEXPR Save the current program to the named file. new Erase the program in memory. All files are closed and all variables are cleared. clear All variables are cleared. All arrays and string variables are deallocated. run { LINENUM } run { STRINGEXPR { , LINENUM } } Begin execution of the program at the first line, or at the specified line. All variables are cleared. If a STRINGEXPR is given then the BASIC program with that name file is loaded into memory first. Program lines are executed in line number order. cont CONTinue execution of the program on the next statement after the statement on which the program stopped execution due to a STOP command or an error.