Atf5 As a Regulator of a Mammalian Mitochondrial Unfolded
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Combinatorial Bzip Dimers Display Complex DNA-Binding Specificity Landscapes
Combinatorial bZIP dimers display complex DNA-binding specificity landscapes The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rodriguez-Martinez, Jose A et al. “Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes.” eLife 6 (2017): n. pag. As Published http://dx.doi.org/10.7554/eLife.19272 Publisher eLife Sciences Publications, Ltd. Version Final published version Citable link http://hdl.handle.net/1721.1/110147 Terms of Use Creative Commons Attribution 4.0 International License Detailed Terms http://creativecommons.org/licenses/by-nc/4.0/ RESEARCH ARTICLE Combinatorial bZIP dimers display complex DNA-binding specificity landscapes Jose´ A Rodrı´guez-Martı´nez1†, Aaron W Reinke2†, Devesh Bhimsaria1,3†, Amy E Keating2,4, Aseem Z Ansari1,5* 1Department of Biochemistry, University of Wisconsin-Madison, Madison, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Electrical and Computer Engineering, University of Wisconsin-Madison, Madison, Unites States; 4Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, United States; 5The Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, United States Abstract How transcription factor dimerization impacts DNA-binding specificity is poorly understood. Guided by protein dimerization properties, we examined DNA binding specificities of 270 human bZIP pairs. DNA interactomes of 80 heterodimers and 22 homodimers revealed that 72% of heterodimer motifs correspond to conjoined half-sites preferred by partnering monomers. Remarkably, the remaining motifs are composed of variably-spaced half-sites (12%) or ‘emergent’ sites (16%) that cannot be readily inferred from half-site preferences of partnering monomers. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Bone Morphogenetic Protein-4 Affects Both Trophoblast and Non-Trophoblast Lineage-Associated Gene Expression in Human Embryonic Stem Cells
Vol.2, No.4, 163-175 (2012) Stem Cell Discovery http://dx.doi.org/10.4236/scd.2012.24021 Bone morphogenetic protein-4 affects both trophoblast and non-trophoblast lineage-associated gene expression in human embryonic stem cells Margaret L. Shirley1,2*, Alison Venable1*, Raj R. Rao3, Nolan L. Boyd4, Steven L. Stice1,5,6, David Puett1#, Prema Narayan7# 1Department of Biochemistry and Molecular Biology, University of Georgia, Athens, USA; #Corresponding Author: [email protected] 2Department of Psychiatry, University of California, San Francisco, USA 3Department of Chemical and Life Science Engineering, School of Engineering, Virginia Commonwealth University, Richmond, USA 4Cardiovascular Innovation Institute, University of Louisville, Louisville, USA 5Regenerative Bioscience Center, University of Georgia, Athens, USA 6Department of Animal and Dairy Sciences, University of Georgia, Athens, USA 7Department of Physiology, Southern Illinois University School of Medicine, Carbondale, USA; #Corresponding Author: [email protected] Received 5 May 2012; revised 4 June 2012; accepted 1 July 2012 ABSTRACT cells were obtained. Gene expression by EB was characterized by an up-regulation of a num- Human embryonic stem cells (hESC) can be in- ber of genes associated with trophoblast, ecto- duced to differentiate to trophoblast by bone derm, endoderm, and mesoderm, and the pro- morphogenetic proteins (BMPs) and by aggre- duction of hCG and progesterone confirmed that gation to form embryoid bodies (EB), but there trophoblast-like cells were formed. These re- are many differences and controversies regard- sults suggest that, in the presence of FGF-2, ing the nature of the differentiated cells. Our BG02 cells respond to BMP4 to yield tropho- goals herein were to determine if BG02 cells form trophoblast-like cells (a) in the presence of blast-like cells, which are also obtained upon EB BMP4-plus-basic fibroblast growth factor (FGF-2) formation. -

Xo GENE PANEL
xO GENE PANEL Targeted panel of 1714 genes | Tumor DNA Coverage: 500x | RNA reads: 50 million Onco-seq panel includes clinically relevant genes and a wide array of biologically relevant genes Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 -

Global Gene Expression Profiling of Oral Cavity Cancers Suggests
BMC Research Notes BioMed Central Short Report Open Access Global gene expression profiling of oral cavity cancers suggests molecular heterogeneity within anatomic subsites Patricia Severino1, Adriana M Alvares2, Pedro Michaluart Jr3, Oswaldo K Okamoto4, Fabio D Nunes5, Carlos A Moreira-Filho6, Eloiza H Tajara*7,8 and Head and Neck Genome Project GENCAPO Address: 1Centro de Pesquisa Experimental, Instituto Israelita de Ensino e Pesquisa Albert Einstein, São Paulo, SP, Brazil, 2Laboratório de Biologia Molecular, Hospital Heliópolis, São Paulo, SP, Brazil, 3Departamento de Cirurgia de Cabeça e Pescoço do Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, São Paulo, SP, Brazil, 4Departamento de Neurologia e Neurocirurgia, Universidade Federal de São Paulo, São Paulo, SP, Brazil, 5Departamento de Estomatologia, Faculdade de Odontologia da Universidade de São Paulo, São Paulo, SP, Brazil, 6Departamento de Pediatria, Faculdade de Medicina da Universidade de São Paulo, São Paulo, SP, Brazil, 7Departamento de Biologia Molecular, Faculdade de Medicina de São José do Rio Preto, São José do Rio Preto, SP, Brazil and 8Departamento de Genética e Biologia Evolutiva, Instituto de Biociências da Universidade de São Paulo, São Paulo, SP, Brazil Email: Patricia Severino - [email protected]; Adriana M Alvares - [email protected]; Pedro Michaluart - [email protected]; [email protected]; [email protected]; Carlos A Moreira-Filho - [email protected]; Eloiza H Tajara* - [email protected]; Head and Neck Genome Project GENCAPO - [email protected] * Corresponding author Published: 13 November 2008 Received: 24 June 2008 Accepted: 13 November 2008 BMC Research Notes 2008, 1:113 doi:10.1186/1756-0500-1-113 This article is available from: http://www.biomedcentral.com/1756-0500/1/113 © 2008 Tajara et al; licensee BioMed Central Ltd. -

Application of Microrna Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease
Article Application of microRNA Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease Jennifer L. Major, Rushita A. Bagchi * and Julie Pires da Silva * Department of Medicine, Division of Cardiology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; [email protected] * Correspondence: [email protected] (R.A.B.); [email protected] (J.P.d.S.) Supplementary Tables Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 www.mdpi.com/journal/mps Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 2 of 25 Table 1. List of all hsa-miRs identified by Human microRNA Disease Database (HMDD; v3.2) analysis. hsa-miRs were identified using the term “genetics” and “circulating” as input in HMDD. Targets CAD hsa-miR-1 Targets IR injury hsa-miR-423 Targets Obesity hsa-miR-499 hsa-miR-146a Circulating Obesity Genetics CAD hsa-miR-423 hsa-miR-146a Circulating CAD hsa-miR-149 hsa-miR-499 Circulating IR Injury hsa-miR-146a Circulating Obesity hsa-miR-122 Genetics Stroke Circulating CAD hsa-miR-122 Circulating Stroke hsa-miR-122 Genetics Obesity Circulating Stroke hsa-miR-26b hsa-miR-17 hsa-miR-223 Targets CAD hsa-miR-340 hsa-miR-34a hsa-miR-92a hsa-miR-126 Circulating Obesity Targets IR injury hsa-miR-21 hsa-miR-423 hsa-miR-126 hsa-miR-143 Targets Obesity hsa-miR-21 hsa-miR-223 hsa-miR-34a hsa-miR-17 Targets CAD hsa-miR-223 hsa-miR-92a hsa-miR-126 Targets IR injury hsa-miR-155 hsa-miR-21 Circulating CAD hsa-miR-126 hsa-miR-145 hsa-miR-21 Targets Obesity hsa-mir-223 hsa-mir-499 hsa-mir-574 Targets IR injury hsa-mir-21 Circulating IR injury Targets Obesity hsa-mir-21 Targets CAD hsa-mir-22 hsa-mir-133a Targets IR injury hsa-mir-155 hsa-mir-21 Circulating Stroke hsa-mir-145 hsa-mir-146b Targets Obesity hsa-mir-21 hsa-mir-29b Methods Protoc. -

Supplementary Material Computational Prediction of SARS
Supplementary_Material Computational prediction of SARS-CoV-2 encoded miRNAs and their putative host targets Sheet_1 List of potential stem-loop structures in SARS-CoV-2 genome as predicted by VMir. Rank Name Start Apex Size Score Window Count (Absolute) Direct Orientation 1 MD13 2801 2864 125 243.8 61 2 MD62 11234 11286 101 211.4 49 4 MD136 27666 27721 104 205.6 119 5 MD108 21131 21184 110 204.7 210 9 MD132 26743 26801 119 188.9 252 19 MD56 9797 9858 128 179.1 59 26 MD139 28196 28233 72 170.4 133 28 MD16 2934 2974 76 169.9 71 43 MD103 20002 20042 80 159.3 403 46 MD6 1489 1531 86 156.7 171 51 MD17 2981 3047 131 152.8 38 87 MD4 651 692 75 140.3 46 95 MD7 1810 1872 121 137.4 58 116 MD140 28217 28252 72 133.8 62 122 MD55 9712 9758 96 132.5 49 135 MD70 13171 13219 93 130.2 131 164 MD95 18782 18820 79 124.7 184 173 MD121 24086 24135 99 123.1 45 176 MD96 19046 19086 75 123.1 179 196 MD19 3197 3236 76 120.4 49 200 MD86 17048 17083 73 119.8 428 223 MD75 14534 14600 137 117 51 228 MD50 8824 8870 94 115.8 79 234 MD129 25598 25642 89 115.6 354 Reverse Orientation 6 MR61 19088 19132 88 197.8 271 10 MR72 23563 23636 148 188.8 286 11 MR11 3775 3844 136 185.1 116 12 MR94 29532 29582 94 184.6 271 15 MR43 14973 15028 109 183.9 226 27 MR14 4160 4206 89 170 241 34 MR35 11734 11792 111 164.2 37 52 MR5 1603 1652 89 152.7 118 53 MR57 18089 18132 101 152.7 139 94 MR8 2804 2864 122 137.4 38 107 MR58 18474 18508 72 134.9 237 117 MR16 4506 4540 72 133.8 311 120 MR34 10010 10048 82 132.7 245 133 MR7 2534 2578 90 130.4 75 146 MR79 24766 24808 75 127.9 59 150 MR65 21528 21576 99 127.4 83 180 MR60 19016 19049 70 122.5 72 187 MR51 16450 16482 75 121 363 190 MR80 25687 25734 96 120.6 75 198 MR64 21507 21544 70 120.3 35 206 MR41 14500 14542 84 119.2 94 218 MR84 26840 26894 108 117.6 94 Sheet_2 List of stable stem-loop structures based on MFE. -

Gene Regulatory Networks Underlying Human Microglia Maturation 2 3 Claudia Z
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.02.446636; this version posted June 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Gene regulatory networks underlying human microglia maturation 2 3 Claudia Z. Han1*, Rick Z. Li1*, Emily Hansen2,3, Hunter R. Bennett1 , Olivier Poirion4, Justin 4 Buchanan1,4, Jean F. Challacombe1, Bethany R. Fixsen1, Samantha Trescott2,3, Johannes C.M. 5 Schlachetzki1, Sebastian Preissl1,4, Allen Wang1,4, Carolyn O’Connor5, Anna S. Warden1,2,3, 6 Shreya Shriram2,3 , Roy Kim2,3, Celina T. Nguyen1,2,3, Danielle M. Schafer2,3, Gabriela Ramirez2,3, 7 Samuel A. Anavim2,3, Avalon Johnson2,3, Eniko Sajti2, Mihir Gupta6, Michael L. Levy6, Sharona 8 Ben-Haim6, David D. Gonda6, Louise Laurent7, Christopher K. Glass1, Nicole G. Coufal2,3,5 9 10 1 Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, 11 CA 92093, USA. 12 2 Department of Pediatrics, University of California, San Diego, La Jolla, CA 92093 13 3 Sanford Consortium for Regenerative Medicine 14 4 Center for Epigenomics, University of California, San Diego, La Jolla, CA 92093 15 5 Salk Institute for Biological Studies, La Jolla, CA 92037 16 6 Department of Neurosurgery, University of California, San Diego-Rady Children's Hospital, San 17 Diego, CA 92123, USA 18 7 Department of Obstetrics, Gynecology, and Reproductive Sciences, University of California, 19 San Diego, CA 92123 20 *These authors contributed equally to this work. -
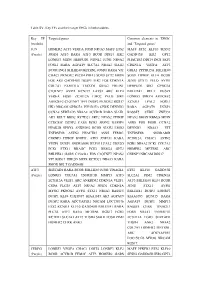
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

A Single-Cell Transcriptome Atlas of the Mouse Glomerulus
RAPID COMMUNICATION www.jasn.org A Single-Cell Transcriptome Atlas of the Mouse Glomerulus Nikos Karaiskos,1 Mahdieh Rahmatollahi,2 Anastasiya Boltengagen,1 Haiyue Liu,1 Martin Hoehne ,2 Markus Rinschen,2,3 Bernhard Schermer,2,4,5 Thomas Benzing,2,4,5 Nikolaus Rajewsky,1 Christine Kocks ,1 Martin Kann,2 and Roman-Ulrich Müller 2,4,5 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Three different cell types constitute the glomerular filter: mesangial depending on cell location relative to the cells, endothelial cells, and podocytes. However, to what extent cellular heteroge- glomerular vascular pole.3 Because BP ad- neity exists within healthy glomerular cell populations remains unknown. aptation and mechanoadaptation of glo- merular cells are key determinants of kidney Methods We used nanodroplet-based highly parallel transcriptional profiling to function and dysregulated in kidney disease, characterize the cellular content of purified wild-type mouse glomeruli. we tested whether glomerular cell type sub- Results Unsupervised clustering of nearly 13,000 single-cell transcriptomes identi- sets can be identified by single-cell RNA fied the three known glomerular cell types. We provide a comprehensive online sequencing in wild-type glomeruli. This atlas of gene expression in glomerular cells that can be queried and visualized using technique allows for high-throughput tran- an interactive and freely available database. Novel marker genes for all glomerular scriptome profiling of individual cells and is cell types were identified and supported by immunohistochemistry images particularly suitable for identifying novel obtained from the Human Protein Atlas.