Jaguar User Manual
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Density Functional Theory
Density Functional Approach Francesco Sottile Ecole Polytechnique, Palaiseau - France European Theoretical Spectroscopy Facility (ETSF) 22 October 2010 Density Functional Theory 1. Any observable of a quantum system can be obtained from the density of the system alone. < O >= O[n] Hohenberg, P. and W. Kohn, 1964, Phys. Rev. 136, B864 Density Functional Theory 1. Any observable of a quantum system can be obtained from the density of the system alone. < O >= O[n] 2. The density of an interacting-particles system can be calculated as the density of an auxiliary system of non-interacting particles. Hohenberg, P. and W. Kohn, 1964, Phys. Rev. 136, B864 Kohn, W. and L. Sham, 1965, Phys. Rev. 140, A1133 Density Functional ... Why ? Basic ideas of DFT Importance of the density Example: atom of Nitrogen (7 electron) 1. Any observable of a quantum Ψ(r1; ::; r7) 21 coordinates system can be obtained from 10 entries/coordinate ) 1021 entries the density of the system alone. 8 bytes/entry ) 8 · 1021 bytes 4:7 × 109 bytes/DVD ) 2 × 1012 DVDs 2. The density of an interacting-particles system can be calculated as the density of an auxiliary system of non-interacting particles. Density Functional ... Why ? Density Functional ... Why ? Density Functional ... Why ? Basic ideas of DFT Importance of the density Example: atom of Oxygen (8 electron) 1. Any (ground-state) observable Ψ(r1; ::; r8) 24 coordinates of a quantum system can be 24 obtained from the density of the 10 entries/coordinate ) 10 entries 8 bytes/entry ) 8 · 1024 bytes system alone. 5 · 109 bytes/DVD ) 1015 DVDs 2. -

Open Babel Documentation Release 2.3.1
Open Babel Documentation Release 2.3.1 Geoffrey R Hutchison Chris Morley Craig James Chris Swain Hans De Winter Tim Vandermeersch Noel M O’Boyle (Ed.) December 05, 2011 Contents 1 Introduction 3 1.1 Goals of the Open Babel project ..................................... 3 1.2 Frequently Asked Questions ....................................... 4 1.3 Thanks .................................................. 7 2 Install Open Babel 9 2.1 Install a binary package ......................................... 9 2.2 Compiling Open Babel .......................................... 9 3 obabel and babel - Convert, Filter and Manipulate Chemical Data 17 3.1 Synopsis ................................................. 17 3.2 Options .................................................. 17 3.3 Examples ................................................. 19 3.4 Differences between babel and obabel .................................. 21 3.5 Format Options .............................................. 22 3.6 Append property values to the title .................................... 22 3.7 Filtering molecules from a multimolecule file .............................. 22 3.8 Substructure and similarity searching .................................. 25 3.9 Sorting molecules ............................................ 25 3.10 Remove duplicate molecules ....................................... 25 3.11 Aliases for chemical groups ....................................... 26 4 The Open Babel GUI 29 4.1 Basic operation .............................................. 29 4.2 Options ................................................. -

The Alexandria Library, a Quantum-Chemical Database of Molecular Properties for Force field Development 9 2017 Received: October 1 1 1 Mohammad M
www.nature.com/scientificdata OPEN Data Descriptor: The Alexandria library, a quantum-chemical database of molecular properties for force field development 9 2017 Received: October 1 1 1 Mohammad M. Ghahremanpour , Paul J. van Maaren & David van der Spoel Accepted: 19 February 2018 Published: 10 April 2018 Data quality as well as library size are crucial issues for force field development. In order to predict molecular properties in a large chemical space, the foundation to build force fields on needs to encompass a large variety of chemical compounds. The tabulated molecular physicochemical properties also need to be accurate. Due to the limited transparency in data used for development of existing force fields it is hard to establish data quality and reusability is low. This paper presents the Alexandria library as an open and freely accessible database of optimized molecular geometries, frequencies, electrostatic moments up to the hexadecupole, electrostatic potential, polarizabilities, and thermochemistry, obtained from quantum chemistry calculations for 2704 compounds. Values are tabulated and where available compared to experimental data. This library can assist systematic development and training of empirical force fields for a broad range of molecules. Design Type(s) data integration objective • molecular physical property analysis objective Measurement Type(s) physicochemical characterization Technology Type(s) Computational Chemistry Factor Type(s) Sample Characteristic(s) 1 Uppsala Centre for Computational Chemistry, Science for Life Laboratory, Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, SE-75124 Uppsala, Sweden. Correspondence and requests for materials should be addressed to D.v.d.S. (email: [email protected]). -
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Camcasp 5.9 Alston J. Misquitta† and Anthony J
CamCASP 5.9 Alston J. Misquittay and Anthony J. Stoneyy yDepartment of Physics and Astronomy, Queen Mary, University of London, 327 Mile End Road, London E1 4NS yy University Chemical Laboratory, Lensfield Road, Cambridge CB2 1EW September 1, 2015 Abstract CamCASP is a suite of programs designed to calculate molecular properties (multipoles and frequency- dependent polarizabilities) in single-site and distributed form, and interaction energies between pairs of molecules, and thence to construct atom–atom potentials. The CamCASP distribution also includes the programs Pfit, Casimir,Gdma 2.2, Cluster, and Process. Copyright c 2007–2014 Alston J. Misquitta and Anthony J. Stone Contents 1 Introduction 1 1.1 Authors . .1 1.2 Citations . .1 2 What’s new? 2 3 Outline of the capabilities of CamCASP and other programs 5 3.1 CamCASP limits . .7 4 Installation 7 4.1 Building CamCASP from source . .9 5 Using CamCASP 10 5.1 Workflows . 10 5.2 High-level scripts . 10 5.3 The runcamcasp.py script . 11 5.4 Low-level scripts . 13 6 Data conventions 13 7 CLUSTER: Detailed specification 14 7.1 Prologue . 15 7.2 Molecule definitions . 15 7.3 Geometry manipulations and other transformations . 16 7.4 Job specification . 18 7.5 Energy . 23 7.6 Crystal . 25 7.7 ORIENT ............................................... 26 7.8 Finally, . 29 8 Examples 29 8.1 A SAPT(DFT) calculation . 29 8.2 An example properties calculation . 30 8.3 Dispersion coefficients . 34 8.4 Using CLUSTER to obtain the dimer geometry . 36 9 CamCASP program specification 39 9.1 Global data . -

Designing Universal Chemical Markup (UCM) Through the Reusable Methodology Based on Analyzing Existing Related Formats
Designing Universal Chemical Markup (UCM) through the reusable methodology based on analyzing existing related formats Background: In order to design concepts for a new general-purpose chemical format we analyzed the strengths and weaknesses of current formats for common chemical data. While the new format is discussed more in the next article, here we describe our software s t tools and two stage analysis procedure that supplied the necessary information for the n i r development. The chemical formats analyzed in both stages were: CDX, CDXML, CML, P CTfile and XDfile. In addition the following formats were included in the first stage only: e r P CIF, InChI, NCBI ASN.1, NCBI XML, PDB, PDBx/mmCIF, PDBML, SMILES, SLN and Mol2. Results: A two stage analysis process devised for both XML (Extensible Markup Language) and non-XML formats enabled us to verify if and how potential advantages of XML are utilized in the widely used general-purpose chemical formats. In the first stage we accumulated information about analyzed formats and selected the formats with the most general-purpose chemical functionality for the second stage. During the second stage our set of software quality requirements was used to assess the benefits and issues of selected formats. Additionally, the detailed analysis of XML formats structure in the second stage helped us to identify concepts in those formats. Using these concepts we came up with the concise structure for a new chemical format, which is designed to provide precise built-in validation capabilities and aims to avoid the potential issues of analyzed formats. -

Notes on OLEX2
Notes on OLEX2 Updated on 12 January 2018,at 09:05. Olex2 v1.2-dev © OlexSys Ltd. 2004 – 2016 Compilation Info: 2017.07.20 svn.r3457 MSC:150030729 on WIN64, Python: 2.7.5, wxWidgets: 3.1.0 for OlexSys Ilia A. Guzei 2124 Chemistry Department, University of Wisconsin-Madison, 1101 University Ave, Madison, WI 53706 USA. This is work in progress. You are encouraged to e-mail me ([email protected]) your comments, corrections, and suggestions. Many thanks to Nattamai Bhuvanesh, Brian Dolinar, Oleg Dolomanov, Dean Johnston, Horst Puschmann, Amy Sarjeant, Charlotte Stern, for proof- reading, suggestions, and comments. I have also borrowed from Martin Lutz, Len Barbour, Richard Staples and Tony Linden. OLEX2 Manual Table of Content Table of Content ........................................................................................................................... 2 How to install OLEX2 under Windows .......................................................................................... 3 How to install OLEX2 on a Mac .................................................................................................... 6 Installing and using PLATON on a Mac ........................................................................................ 8 How to get OLEX2 to use PLATON ............................................................................................ 11 About program OLEX2 ................................................................................................................ 11 Keyboard shortcuts ..................................................................................................................... -

Visualizing 3D Molecular Structures Using an Augmented Reality App
Visualizing 3D molecular structures using an augmented reality app Kristina Eriksen, Bjarne E. Nielsen, Michael Pittelkow 5 Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark. E-mail: [email protected] ABSTRACT 10 We present a simple procedure to make an augmented reality app to visualize any 3D chemical model. The molecular structure may be based on data from crystallographic data or from computer modelling. This guide is made in such a way, that no programming skills are needed and the procedure uses free software and is a way to visualize 3D structures that are normally difficult to comprehend in the 2D 15 space of paper. The process can be applied to make 3D representation of any 2D object, and we envisage the app to be useful when visualizing simple stereochemical problems, when presenting a complex 3D structure on a poster presentation or even in audio-visual presentations. The method works for all molecules including small molecules, supramolecular structures, MOFs and biomacromolecules. GRAPHICAL ABSTRACT 20 KEYWORDS Augmented reality, Unity, Vuforia, Application, 3D models. 25 Journal 5/18/21 Page 1 of 14 INTRODUCTION Conveying information about three-dimensional (3D) structures in two-dimensional (2D) space, such as on paper or a screen can be difficult. Augmented reality (AR) provides an opportunity to visualize 2D 30 structures in 3D. Software to make simple AR apps is becoming common and ranges of free software now exist to make customized apps. AR has transformed visualization in computer games and films, but the technique is distinctly under-used in (chemical) science.1 In chemical science the challenge of visualizing in 3D exists at several levels ranging from teaching of stereo chemistry problems at freshman university level to visualizing complex molecular structures at 35 the forefront of chemical research. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -
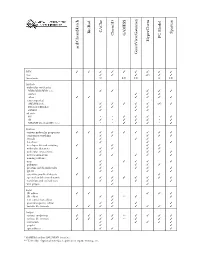
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Dr. David Danovich
Dr. David Danovich ! 14 January 2021 (h-index: 34; 124 publications) Personal Information Name: Contact information Danovich David Private: Nissan Harpaz st., 4/9, Jerusalem, Date of Birth: 9371643 Israel June 29, 1959 Mobile: +972-54-4768669 E-mail: [email protected] Place of Birth: Irkutsk, USSR Work: Institute of Chemistry, The Hebrew Citizenship: University, Edmond Safra Campus, Israeli Givat Ram, 9190401 Jerusalem, Israel Marital status: Phone: +972-2-6586934 Married +2 FAX: +972-2-6584033 E-mail: [email protected] Current position 1992- present: Senior computational chemist and scientific programmer in the Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel Scientific skills Very experienced scientific researcher with demonstrated ability to implement ab initio molecular electronic structure and valence bond calculations for obtaining insights into physical and chemical properties of molecules and materials. Extensive expertise in implementation of Molecular Orbital Theory based on ab initio methods such as Hartree-Fock theory (HF), Density Functional Theory (DFT), Moller-Plesset Perturbation theory (MP2-MP5), Couple Cluster theories [CCST(T)], Complete Active Space (CAS, CASPT2) theories, Multireference Configuration Interaction (MRCI) theory and modern ab initio valence bond theory (VBT) methods such as Valence Bond Self Consistent Field theory (VBSCF), Breathing Orbital Valence Bond theory (BOVB) and its variants (like D-BOVB, S-BOVB and SD-BOVB), Valence Bond Configuration Interaction theory (VBCI), Valence Bond Perturbation theory (VBPT). Language skills Advanced Conversational English, Russian Hebrew Academic background 1. Undergraduate studies, 1978-1982 Master of Science in Physics, June 1982 Irkutsk State University, USSR Dr. David Danovich ! 2. Graduate studies, 1984-1989 Ph. -
![Arxiv:1710.00259V1 [Physics.Chem-Ph] 30 Sep 2017 a Relativistically Correct Electron Interaction](https://docslib.b-cdn.net/cover/2561/arxiv-1710-00259v1-physics-chem-ph-30-sep-2017-a-relativistically-correct-electron-interaction-842561.webp)
Arxiv:1710.00259V1 [Physics.Chem-Ph] 30 Sep 2017 a Relativistically Correct Electron Interaction
One-step treatment of spin-orbit coupling and electron correlation in large active spaces Bastien Mussard1, ∗ and Sandeep Sharma1, y 1Department of Chemistry and Biochemistry, University of Colorado Boulder, Boulder, CO 80302, USA In this work we demonstrate that the heat bath configuration interaction (HCI) and its semis- tochastic extension can be used to treat relativistic effects and electron correlation on an equal footing in large active spaces to calculate the low energy spectrum of several systems including halogens group atoms (F, Cl, Br, I), coinage atoms (Cu, Au) and the Neptunyl(VI) dioxide radical. This work demonstrates that despite a significant increase in the size of the Hilbert space due to spin symmetry breaking by the spin-orbit coupling terms, HCI retains the ability to discard large parts of the low importance Hilbert space to deliver converged absolute and relative energies. For instance, by using just over 107 determinants we get converged excitation energies for Au atom in an active space containing (150o,25e) which has over 1030 determinants. We also investigate the accuracy of five different two-component relativistic Hamiltonians in which different levels of ap- proximations are made in deriving the one-electron and two-electrons Hamiltonians, ranging from Breit-Pauli (BP) to various flavors of exact two-component (X2C) theory. The relative accuracy of the different Hamiltonians are compared on systems that range in atomic number from first row atoms to actinides. I. INTRODUCTION can become large and it is more appropriate to treat them on an equal footing with the spin-free terms and electron Relativistic effects play an important role in a correlation.