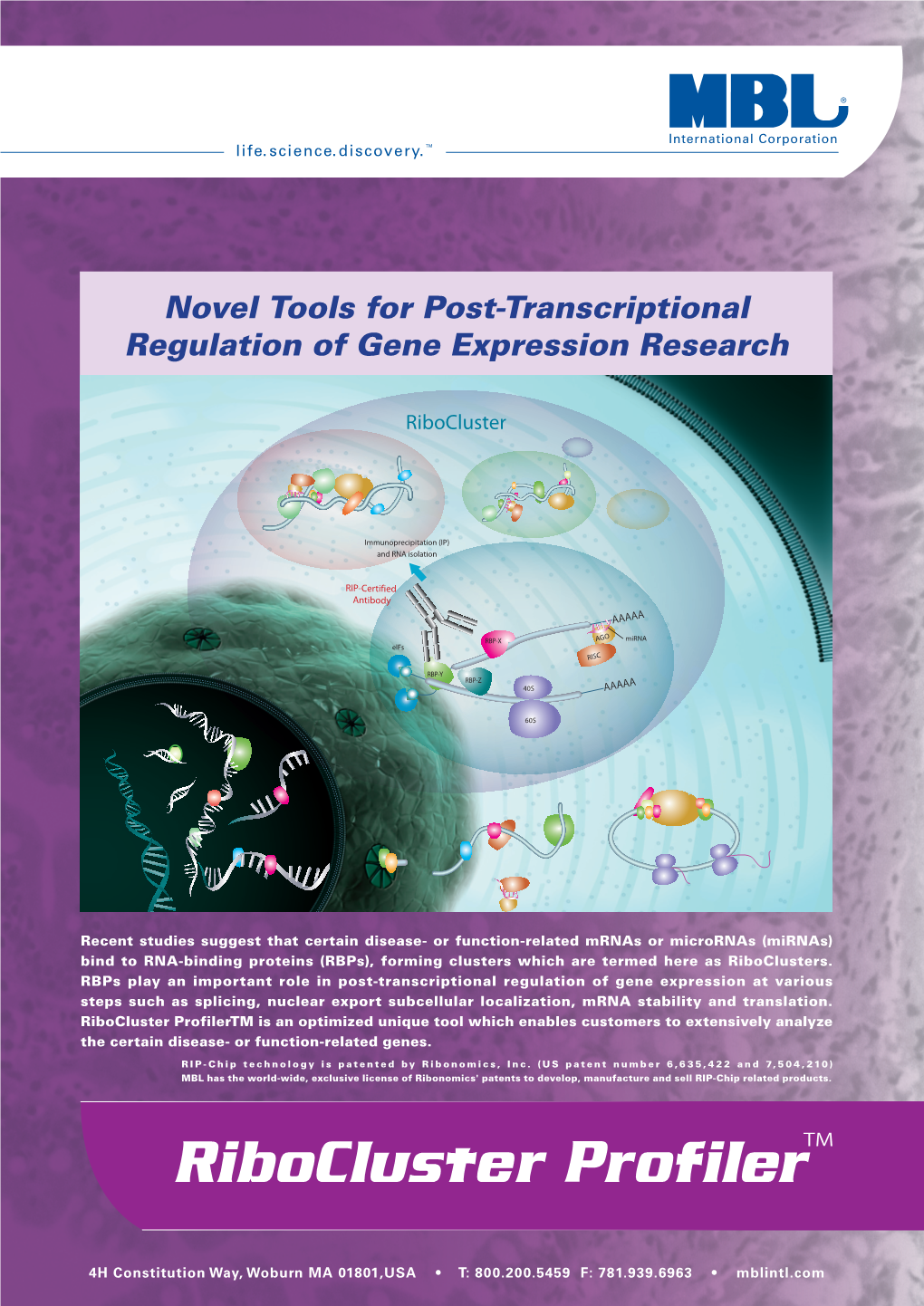
Novel Tools for Post-Transcriptional Regulation of Gene Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Research Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks Endre Sebestyén,1,5 Babita Singh,1,5 Belén Miñana,1,2 Amadís Pagès,1 Francesca Mateo,3 Miguel Angel Pujana,3 Juan Valcárcel,1,2,4 and Eduardo Eyras1,4 1Universitat Pompeu Fabra, E08003 Barcelona, Spain; 2Centre for Genomic Regulation, E08003 Barcelona, Spain; 3Program Against Cancer Therapeutic Resistance (ProCURE), Catalan Institute of Oncology (ICO), Bellvitge Institute for Biomedical Research (IDIBELL), E08908 L’Hospitalet del Llobregat, Spain; 4Catalan Institution for Research and Advanced Studies, E08010 Barcelona, Spain Alternative splicing is regulated by multiple RNA-binding proteins and influences the expression of most eukaryotic genes. However, the role of this process in human disease, and particularly in cancer, is only starting to be unveiled. We system- atically analyzed mutation, copy number, and gene expression patterns of 1348 RNA-binding protein (RBP) genes in 11 solid tumor types, together with alternative splicing changes in these tumors and the enrichment of binding motifs in the alter- natively spliced sequences. Our comprehensive study reveals widespread alterations in the expression of RBP genes, as well as novel mutations and copy number variations in association with multiple alternative splicing changes in cancer drivers and oncogenic pathways. Remarkably, the altered splicing patterns in several tumor types recapitulate those of undifferen- tiated cells. These patterns are predicted to be mainly controlled by MBNL1 and involve multiple cancer drivers, including the mitotic gene NUMA1. We show that NUMA1 alternative splicing induces enhanced cell proliferation and centrosome am- plification in nontumorigenic mammary epithelial cells. -

Sanjay Kumar Gupta
The human CCHC-type Zinc Finger Nucleic Acid Binding Protein (CNBP) binds to the G-rich elements in target mRNA coding sequences and promotes translation Das humane CCHC-Typ-Zinkfinger-Nukleinsäure-Binde-Protein (CNBP) bindet an G-reiche Elemente in der kodierenden Sequenz seiner Ziel-mRNAs und fördert deren Translation Doctoral thesis for a doctoral degree at the Graduate School of Life Sciences, Julius-Maximilians-Universität WürzBurg, Section: Biomedicine suBmitted By Sanjay Kumar Gupta from Varanasi, India WürzBurg, 2016 1 Submitted on: …………………………………………………………..…….. Office stamp Members of the Promotionskomitee: Chairperson: Prof. Dr. Alexander Buchberger Primary Supervisor: Dr. Stefan Juranek Supervisor (Second): Prof. Dr. Utz Fischer Supervisor (Third): Dr. Markus Landthaler Date of Public Defence: …………………………………………….………… Date of Receipt of Certificates: ………………………………………………. 2 Summary The genetic information encoded with in the genes are transcribed and translated to give rise to the functional proteins, which are building block of a cell. At first, it was thought that the regulation of gene expression particularly occurs at the level of transcription By various transcription factors. Recent discoveries have shown the vital role of gene regulation at the level of RNA also known as post-transcriptional gene regulation (PTGR). Apart from non-coding RNAs e.g. micro RNAs, various RNA Binding proteins (RBPs) play essential role in PTGR. RBPs have been implicated in different stages of mRNA life cycle ranging from splicing, processing, transport, localization and decay. In last 20 years studies have shown the presence of hundreds of RBPs across eukaryotic systems many of which are widely conserved. Given the rising numBer of RBPs and their link to human diseases it is quite evident that RBPs have major role in cellular processes and their regulation. -

Differential Analysis of Gene Expression and Alternative Splicing
i bioRxiv preprint doi: https://doi.org/10.1101/2020.08.23.234237; this version posted August 24, 2020. The copyright holder for this preprint i (which was not certified by peer review)“main” is the author/funder, — 2020/8/23 who has — granted 9:38 — bioRxiv page a1 license — #1 to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. i i EmpiReS: Differential Analysis of Gene Expression and Alternative Splicing Gergely Csaba∗, Evi Berchtold*,y, Armin Hadziahmetovic, Markus Gruber, Constantin Ammar and Ralf Zimmer Department of Informatics, Ludwig-Maximilians-Universitat¨ Munchen,¨ 80333, Munich, Germany ABSTRACT to translation. Different transcripts of the same gene are often expressed at the same time, possibly at different abundance, While absolute quantification is challenging in high- yielding a defined mixture of isoforms. Changes to this throughput measurements, changes of features composition are further referred to as differential alternative between conditions can often be determined with splicing (DAS). Various diseases can be linked to DAS high precision. Therefore, analysis of fold changes events (3). Most hallmarks of cancer like tumor progression is the standard method, but often, a doubly and immune escape (4, 5) can be attributed to changes in differential analysis of changes of changes is the transcript composition (6). DAS plays a prominent role required. Differential alternative splicing is an in various types of muscular dystrophy (MD) (7), splicing example of a doubly differential analysis, i.e. fold intervention by targeted exon removal is a therapeutic option changes between conditions for different isoforms in Duchenne MD (8). -

Supplementary Table S1. Upregulated Genes Differentially
Supplementary Table S1. Upregulated genes differentially expressed in athletes (p < 0.05 and 1.3-fold change) Gene Symbol p Value Fold Change 221051_s_at NMRK2 0.01 2.38 236518_at CCDC183 0.00 2.05 218804_at ANO1 0.00 2.05 234675_x_at 0.01 2.02 207076_s_at ASS1 0.00 1.85 209135_at ASPH 0.02 1.81 228434_at BTNL9 0.03 1.81 229985_at BTNL9 0.01 1.79 215795_at MYH7B 0.01 1.78 217979_at TSPAN13 0.01 1.77 230992_at BTNL9 0.01 1.75 226884_at LRRN1 0.03 1.74 220039_s_at CDKAL1 0.01 1.73 236520_at 0.02 1.72 219895_at TMEM255A 0.04 1.72 201030_x_at LDHB 0.00 1.69 233824_at 0.00 1.69 232257_s_at 0.05 1.67 236359_at SCN4B 0.04 1.64 242868_at 0.00 1.63 1557286_at 0.01 1.63 202780_at OXCT1 0.01 1.63 1556542_a_at 0.04 1.63 209992_at PFKFB2 0.04 1.63 205247_at NOTCH4 0.01 1.62 1554182_at TRIM73///TRIM74 0.00 1.61 232892_at MIR1-1HG 0.02 1.61 204726_at CDH13 0.01 1.6 1561167_at 0.01 1.6 1565821_at 0.01 1.6 210169_at SEC14L5 0.01 1.6 236963_at 0.02 1.6 1552880_at SEC16B 0.02 1.6 235228_at CCDC85A 0.02 1.6 1568623_a_at SLC35E4 0.00 1.59 204844_at ENPEP 0.00 1.59 1552256_a_at SCARB1 0.02 1.59 1557283_a_at ZNF519 0.02 1.59 1557293_at LINC00969 0.03 1.59 231644_at 0.01 1.58 228115_at GAREM1 0.01 1.58 223687_s_at LY6K 0.02 1.58 231779_at IRAK2 0.03 1.58 243332_at LOC105379610 0.04 1.58 232118_at 0.01 1.57 203423_at RBP1 0.02 1.57 AMY1A///AMY1B///AMY1C///AMY2A///AMY2B// 208498_s_at 0.03 1.57 /AMYP1 237154_at LOC101930114 0.00 1.56 1559691_at 0.01 1.56 243481_at RHOJ 0.03 1.56 238834_at MYLK3 0.01 1.55 213438_at NFASC 0.02 1.55 242290_at TACC1 0.04 1.55 ANKRD20A1///ANKRD20A12P///ANKRD20A2/// -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Immunoprecipitation and Mass Spectrometry Defines an Extensive
BRES : 44759 Model7 pp: À 1221ðcol:fig: : NILÞ brain research ] ( ]]]]) ]]]– ]]] Available online at www.sciencedirect.com 121 122 123 124 125 126 www.elsevier.com/locate/brainres 127 128 129 Review 130 131 fi 132 Immunoprecipitation and mass spectrometry de nes 133 – 134 an extensive RBM45 protein protein interaction 135 Q2 136 network 137 138 a a,b a a c 139 Yang Li , Mahlon Collins , Jiyan An , Rachel Geiser , Tony Tegeler , c c c a,b,n 140 Q1 Kristine Tsantilas , Krystine Garcia , Patrick Pirrotte , Robert Bowser 141 aDivisions of Neurology and Neurobiology, Barrow Neurological Institute, St. Joseph's Hospital and Medical Center, 142 Phoenix, AZ 85013, USA 143 bUniversity of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA 144 cCenter for Proteomics, TGen (Translational Genomics Research Institute), Phoenix, AZ 85004, USA 145 146 147 article info abstract 148 149 Article history: The pathological accumulation of RNA-binding proteins (RBPs) within inclusion bodies is a 150 Received 30 January 2016 hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration 151 Received in revised form (FTLD). RBP aggregation results in both toxic gain and loss of normal function. Determining 152 25 February 2016 the protein binding partners and normal functions of disease-associated RBPs is necessary 153 Accepted 28 February 2016 to fully understand molecular mechanisms of RBPs in disease. Herein, we characterized 154 the protein–protein interactions (PPIs) of RBM45, a RBP that localizes to inclusions in ALS/ 155 – fi Keywords: FTLD. Using immunoprecipitation coupled to mass spectrometry (IP MS), we identi ed 132 156 fi RBM45 proteins that speci cally interact with RBM45 within HEK293 cells. -

Mrna Turnover Philip Mitchell* and David Tollervey†
320 mRNA turnover Philip Mitchell* and David Tollervey† Nuclear RNA-binding proteins can record pre-mRNA are cotransported to the cytoplasm with the mRNP. These processing events in the structure of messenger proteins may preserve a record of the nuclear history of the ribonucleoprotein particles (mRNPs). During initial rounds of pre-mRNA in the cytoplasmic mRNP structure. This infor- translation, the mature mRNP structure is established and is mation can strongly influence the cytoplasmic fate of the monitored by mRNA surveillance systems. Competition for the mRNA and is used by mRNA surveillance systems that act cap structure links translation and subsequent mRNA as a checkpoint of mRNP integrity, particularly in the identi- degradation, which may also involve multiple deadenylases. fication of premature translation termination codons (PTCs). Addresses Cotransport of nuclear mRNA-binding proteins with mRNA Wellcome Trust Centre for Cell Biology, ICMB, University of Edinburgh, from the nucleus to the cytoplasm (nucleocytoplasmic shut- Kings’ Buildings, Edinburgh EH9 3JR, UK tling) was first observed for the heterogeneous nuclear *e-mail: [email protected] ribonucleoprotein (hnRNP) proteins. Some hnRNP proteins †e-mail: [email protected] are stripped from the mRNA at export [1], but hnRNP A1, Current Opinion in Cell Biology 2001, 13:320–325 A2, E, I and K are all exported (see [2]). Although roles for 0955-0674/01/$ — see front matter these hnRNP proteins in transport and translation have been © 2001 Elsevier Science Ltd. All rights reserved. reported [3•,4•], their affects on mRNA stability have been little studied. More is known about hnRNP D/AUF1 and Abbreviations AREs AU-rich sequence elements another nuclear RNA-binding protein, HuR, which act CBC cap-binding complex antagonistically to modulate the stability of a range of DAN deadenylating nuclease mRNAs containing AU-rich sequence elements (AREs) DSEs downstream sequence elements (reviewed in [2]). -

RNA Components of the Spliceosome Regulate Tissue- and Cancer-Specific Alternative Splicing
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing Heidi Dvinge,1,2,4 Jamie Guenthoer,3 Peggy L. Porter,3 and Robert K. Bradley1,2 1Computational Biology Program, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 2Basic Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 3Human Biology Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA Alternative splicing of pre-mRNAs plays a pivotal role during the establishment and maintenance of human cell types. Characterizing the trans-acting regulatory proteins that control alternative splicing has therefore been the focus of much research. Recent work has established that even core protein components of the spliceosome, which are required for splicing to proceed, can nonetheless contribute to splicing regulation by modulating splice site choice. We here show that the RNA components of the spliceosome likewise influence alternative splicing decisions. Although these small nuclear RNAs (snRNAs), termed U1, U2, U4, U5, and U6 snRNA, are present in equal stoichiometry within the spliceosome, we found that their relative levels vary by an order of magnitude during development, across tissues, and across cancer samples. Physiologically relevant perturbation of individual snRNAs drove widespread gene-specific differences in alternative splic- ing but not transcriptome-wide splicing failure. Genes that were particularly sensitive to variations in snRNA abundance in a breast cancer cell line model were likewise preferentially misspliced within a clinically diverse cohort of invasive breast ductal carcinomas. -

Comparative Interactomics Analysis of Different ALS-Associated Proteins
Acta Neuropathol (2016) 132:175–196 DOI 10.1007/s00401-016-1575-8 ORIGINAL PAPER Comparative interactomics analysis of different ALS‑associated proteins identifies converging molecular pathways Anna M. Blokhuis1 · Max Koppers1,2 · Ewout J. N. Groen1,2,9 · Dianne M. A. van den Heuvel1 · Stefano Dini Modigliani4 · Jasper J. Anink5,6 · Katsumi Fumoto1,10 · Femke van Diggelen1 · Anne Snelting1 · Peter Sodaar2 · Bert M. Verheijen1,2 · Jeroen A. A. Demmers7 · Jan H. Veldink2 · Eleonora Aronica5,6 · Irene Bozzoni3 · Jeroen den Hertog8 · Leonard H. van den Berg2 · R. Jeroen Pasterkamp1 Received: 22 January 2016 / Revised: 14 April 2016 / Accepted: 15 April 2016 / Published online: 10 May 2016 © The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Amyotrophic lateral sclerosis (ALS) is a dev- OPTN and UBQLN2, in which mutations caused loss or astating neurological disease with no effective treatment gain of protein interactions. Several of the identified inter- available. An increasing number of genetic causes of ALS actomes showed a high degree of overlap: shared binding are being identified, but how these genetic defects lead to partners of ATXN2, FUS and TDP-43 had roles in RNA motor neuron degeneration and to which extent they affect metabolism; OPTN- and UBQLN2-interacting proteins common cellular pathways remains incompletely under- were related to protein degradation and protein transport, stood. To address these questions, we performed an inter- and C9orf72 interactors function in mitochondria. To con- actomic analysis to identify binding partners of wild-type firm that this overlap is important for ALS pathogenesis, (WT) and ALS-associated mutant versions of ATXN2, we studied fragile X mental retardation protein (FMRP), C9orf72, FUS, OPTN, TDP-43 and UBQLN2 in neuronal one of the common interactors of ATXN2, FUS and TDP- cells. -

1 Title 1 Loss of PABPC1 Is Compensated by Elevated PABPC4
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.07.430165; this version posted February 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 1 Title 2 Loss of PABPC1 is compensated by elevated PABPC4 and correlates with transcriptome 3 changes 4 5 Jingwei Xie1, 2, Xiaoyu Wei1, Yu Chen1 6 7 1 Department of Biochemistry and Groupe de recherche axé sur la structure des 8 protéines, McGill University, Montreal, Quebec H3G 0B1, Canada 9 10 2 To whom correspondence should be addressed: Dept. of Biochemistry, McGill 11 University, Montreal, QC H3G 0B1, Canada. E-mail: [email protected]. 12 13 14 15 Abstract 16 Cytoplasmic poly(A) binding protein (PABP) is an essential translation factor that binds to 17 the 3' tail of mRNAs to promote translation and regulate mRNA stability. PABPC1 is the 18 most abundant of several PABP isoforms that exist in mammals. Here, we used the 19 CRISPR/Cas genome editing system to shift the isoform composition in HEK293 cells. 20 Disruption of PABPC1 elevated PABPC4 levels. Transcriptome analysis revealed that the 21 shift in the dominant PABP isoform was correlated with changes in key transcriptional 22 regulators. This study provides insight into understanding the role of PABP isoforms in 23 development and differentiation. 24 Keywords 25 PABPC1, PABPC4, c-Myc 26 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.07.430165; this version posted February 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Splicing Graphs and RNA-Seq Data
Splicing graphs and RNA-seq data Herv´ePag`es Daniel Bindreither Marc Carlson Martin Morgan Last modified: January 2019; Compiled: May 19, 2021 Contents 1 Introduction 1 2 Splicing graphs 2 2.1 Definitions and example . .2 2.2 Details about nodes and edges . .2 2.3 Uninformative nodes . .5 3 Computing splicing graphs from annotations 6 3.1 Choosing and loading a gene model . .6 3.2 Generating a SplicingGraphs object . .6 3.3 Basic manipulation of a SplicingGraphs object . .8 3.4 Extracting and plotting graphs from a SplicingGraphs object . 12 4 Splicing graph bubbles 15 4.1 Some definitions . 15 4.2 Computing the bubbles of a splicing graph . 17 4.3 AScodes ............................................... 18 4.4 Tabulating all the AS codes for Human chromosome 14 . 19 5 Counting reads 20 5.1 Assigning reads to the edges of a SplicingGraphs object . 22 5.2 How does read assignment work? . 23 5.3 Counting and summarizing the assigned reads . 25 6 Session Information 26 1 Introduction The SplicingGraphs package allows the user to create and manipulate splicing graphs [1] based on annotations for a given organism. Annotations must describe a gene model, that is, they need to contain the following information: The exact exon/intron structure (i.e., genomic coordinates) for the known transcripts. The grouping of transcripts by gene. Annotations need to be provided as a TxDb or GRangesList object, which is how a gene model is typically represented in Bioconductor. 1 The SplicingGraphs package defines the SplicingGraphs container for storing the splicing graphs together with the gene model that they are based on. -

The Sub-Nuclear Localization of RNA-Binding Proteins in KSHV-Infected Cells
cells Article The Sub-Nuclear Localization of RNA-Binding Proteins in KSHV-Infected Cells Ella Alkalay, Chen Gam Ze Letova Refael, Irit Shoval, Noa Kinor, Ronit Sarid and Yaron Shav-Tal * The Mina & Everard Goodman Faculty of Life Sciences and The Institute of Nanotechnology and Advanced Materials, Bar-Ilan University, Ramat Gan 5290002, Israel; [email protected] (E.A.); [email protected] (C.G.Z.L.R.); [email protected] (I.S.); [email protected] (N.K.); [email protected] (R.S.) * Correspondence: [email protected] Received: 14 August 2020; Accepted: 21 August 2020; Published: 25 August 2020 Abstract: RNA-binding proteins, particularly splicing factors, localize to sub-nuclear domains termed nuclear speckles. During certain viral infections, as the nucleus fills up with replicating virus compartments, host cell chromatin distribution changes, ending up condensed at the nuclear periphery. In this study we wished to determine the fate of nucleoplasmic RNA-binding proteins and nuclear speckles during the lytic cycle of the Kaposi’s sarcoma associated herpesvirus (KSHV). We found that nuclear speckles became fewer and dramatically larger, localizing at the nuclear periphery, adjacent to the marginalized chromatin. Enlarged nuclear speckles contained splicing factors, whereas other proteins were nucleoplasmically dispersed. Polyadenylated RNA, typically found in nuclear speckles under regular conditions, was also found in foci separated from nuclear speckles in infected cells. Poly(A) foci did not contain lncRNAs known to colocalize with nuclear speckles but contained the poly(A)-binding protein PABPN1. Examination of the localization of spliced viral RNAs revealed that some spliced transcripts could be detected within the nuclear speckles.