A Theoretical Study of Carbamate Formation James Ray Diers Master of Science
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Department of Chemistry
ST.PHILOMENA’S COLLEGE (AUTONOMOUS), MYSURU (AFFILIATED TO UNIVERSITY OF MYSORE) REACCREDITED BY NAAC WITH A GRADE COURSE – B.Sc Three-year six semesters Choice Based Credit System (CBCS) and Continuous Assessment & Grading Pattern (CAGP) Under Graduate Programme under Autonomous Structure Academic year 2018-19 onwards DEPARTMENT OF CHEMISTRY St. Philomena’s College (Autonomous) Mysuru. B.Sc., Chemistry Syllabus - CBCS Scheme 2018-19 onwards. Page 1 PREAMBLE For the development of any Society, Science education plays an important role. Chemistry, being a major component of Science, is one of the increasingly important disciplinary areas of Science. Chemistry, which is also studied at the Bachelor’s degree programme for years, has been witnessing a slow transition from an analog to a much- needed one. It is an experimental science and students need to be trained both in the theoretical & practical aspects to get expertise. Moreover, the topics prescribed should provide in-depth knowledge of the subject and also the relevant basic allied subjects. Under this context, in order to make the U. G. teaching more effective and meaningful, revamping the syllabus is the need of the hour. It is certain that systematic and planned curricula from first to the third year shall motivate and encourage students for pursuing higher studies in various disciplines of chemistry such as Inorganic, organic, Physical, Analytical and Bio-Chemistry. This curriculum also enables the students to shoulder the responsibility as chemists in chemical industry. Thus, an updated and content revision of UG Chemistry syllabus is essential to improve its quality at the National and International level and also to meet the present-day challenges of PG and research oriented work after the PG programme. -

Ammonia Snow-Lines and Ammonium Salts Desorption ? F
Astronomy & Astrophysics manuscript no. aanda_ammonium-salts ©ESO 2021 April 22, 2021 Ammonia snow-lines and ammonium salts desorption ? F. Kruczkiewicz1; 2; 3, J. Vitorino2, E. Congiu2, P. Theulé1, and F. Dulieu2 1 Aix Marseille Univ, CNRS, CNES, LAM, Marseille, France e-mail: [email protected] 2 CY Cergy Paris Université, Observatoire de Paris, PSL University, Sorbonne Université, CNRS, LERMA, F-95000, Cergy, France 3 Max-Planck-Institut für extraterrestrische Physik, Gießenbachstraße 1, Garching, 85748, Germany Received –; accepted – ABSTRACT Context. The nitrogen reservoir in planetary systems is a long standing problem. Part of the N-bearing molecules is probably incor- porated into the ice bulk during the cold phases of the stellar evolution, and may be gradually released into the gas phase when the ice is heated, such as in active comets. The chemical nature of the N-reservoir should greatly influence how, when and in what form N returns to the gas phase, or is incorporated into the refractory material forming planetary bodies. Aims. We present the study the thermal desorption of two ammonium salts: ammonium formate and ammonium acetate from a gold surface and from a water ice substrate. Methods. Temperature-programmed desorption experiments and Fourier transform infrared reflection spectroscopy were conducted to investigate the desorption behavior of ammonium salts. Results. Ammonium salts are semi-volatile species releasing neutral species as major components upon desorption, that is ammonia and the corresponding organic acid (HCOOH and CH3COOH), at temperatures higher than the temperature of thermal desorption of water ice. Their desorption follows a first-order Wigner-Polanyi law. We find the first order kinetic parameters A = 7.7 ± 0.6 × 1015 s−1 −1 20 −1 −1 and Ebind = 68.9 ± 0.1 kJ mol for ammonium formate and A = 3.0 ± 0.4 × 10 s and Ebind = 83.0 ± 0.2 kJ mol for ammonium acetate. -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Amino Acid Metabolism, Urea Cycle Semester Iv, Paper Cc8
AMINO ACID METABOLISM, UREA CYCLE SEMESTER IV, PAPER CC8 UREA CYCLE + Some of the NH4 formed in the breakdown of amino acids is consumed in the biosynthesis of nitrogen compounds. Most aquatic species, such as the bony fishes, are ammonotelic, excreting amino nitrogen as ammonia. The toxic ammonia is simply diluted in the surrounding water. Most terrestrial animals are ureotelic, excreting amino nitrogen in the form of urea; birds and reptiles are uricotelic, excreting amino nitrogen as uric acid. In ureotelic organisms, the ammonia deposited in the mitochondria of hepatocytes is converted to urea in the urea cycle. This pathway was discovered in 1932 by Hans Krebs. The urea cycle begins inside liver mitochondria, but three of the subsequent steps take place in the cytosol. The first amino group to enter the urea cycle is derived from ammonia in the mitochondrial matrix arising from various amino acids. The urea cycle begins with the coupling of free NH4 + with HCO3- to form carbamoyl phosphate. The synthesis of carbamoyl phosphate, though a simple molecule, is complex, comprising three steps, all catalyzed by carbamoyl phosphate synthetase. - The reaction begins with the phosphorylation of HCO3 to form carboxyphosphate, which then reacts with ammonium ion to form carbamic acid. Finally, a second molecule of ATP phosphorylates carbamic acid to carbamoyl phosphate. The consumption of two molecules of ATP makes this synthesis of carbamoyl phosphate essentially irreversible. Next, the carbamoyl group of carbamoyl phosphate, which has a high transfer potential because of its anhydride bond, is transferred to ornithine to form citrulline, in a reaction catalyzed by + ornithine transcarbamoylase. -

Flight Through the Comet Chury's Dust Cloud Resolves Chemical Mystery 21 January 2020
Flight through the comet Chury's dust cloud resolves chemical mystery 21 January 2020 em. Hans Balsiger, was on board. A key finding from the measurements taken by this instrument was that there appeared to be a lack of nitrogen in Halley's coma—the nebulous covering of comets which forms when a comet passes close to the sun. Although nitrogen (N) was discovered in the form of ammonia (NH3) and hydrocyanic acid (HCN), the incidence was far removed from the expected cosmic incidence. More than 30 years later, researchers have solved this mystery thanks to a happy accident. This is a result of the analysis of data from the Bernese mass spectrometer ROSINA, which collected data on the comet 67P/Churyumov-Gerasimenko, called Chury for short, on board the ESA space probe Rosetta (see info box below). Risky flight through the comet Chury's dust cloud Less than a month before the end of the Rosetta A plume of dust from Comet mission, the space probe was just 1.9 km above 67P/Churyumov–Gerasimenko, seen by the OSIRIS Wide Angle Camera on ESA's Rosetta spacecraft on 3 the surface of Chury as it flew through a dust cloud July 2016. The shadow of the plume is cast across the from the comet. This resulted in a direct impact of basin, which is in the Imhotep region. Credit: dust in the ion source of the mass spectrometer ESA/Rosetta/MPS for OSIRIS Team ROSINA-DFMS (Rosetta Orbiter Sensor for Ion and MPS/UPD/LAM/IAA/SSO/INTA/UPM/DASP/IDA Neutral Analysis-Double Focusing Mass Spectrometer), led by the University of Bern. -

Ammonium Carbamate Hazard Summary Identification
Common Name: AMMONIUM CARBAMATE CAS Number: 1111-78-0 RTK Substance number: 0091 DOT Number: NA 9083 Date: January 1996 Revision: March 2002 ------------------------------------------------------------------------- ------------------------------------------------------------------------- HAZARD SUMMARY WORKPLACE EXPOSURE LIMITS * Ammonium Carbamate can affect you when breathed in. The following exposure limits are for Ammonia: * Exposure can irritate the skin and eyes causing redness and tearing. OSHA: The legal airborne permissible exposure limit * Breathing Ammonium Carbamate can irritate the nose, (PEL) is 50 ppm averaged over an 8-hour throat and lungs causing coughing and/or shortness of workshift. breath. NIOSH: The recommended airborne exposure limit is IDENTIFICATION 25 ppm averaged over a 10-hour workshift and Ammonium Carbamate is a white crystalline (sand-like) 35 ppm, not to be exceeded during any 15 minute powder. It is used as a fertilizer and ammoniating agent. work period. REASON FOR CITATION ACGIH: The recommended airborne exposure limit is 25 ppm averaged over an 8-hour workshift and * Ammonium Carbamate is on the Hazardous Substance 35 ppm as a STEL (short-term exposure limit). List because it is cited by DOT and EPA. * Definitions are provided on page 5. WAYS OF REDUCING EXPOSURE HOW TO DETERMINE IF YOU ARE BEING * Where possible, enclose operations and use local exhaust ventilation at the site of chemical release. If local exhaust EXPOSED ventilation or enclosure is not used, respirators should be The New Jersey Right to Know Act requires most employers worn. to label chemicals in the workplace and requires public * Wear protective work clothing. employers to provide their employees with information and * Wash thoroughly immediately after exposure to training concerning chemical hazards and controls. -

TR-541: Formamide (CASRN 75-12-7) in F344/N Rats and B6C3F1 Mice (Gavage Studies)
NTP TECHNICAL REPORT ON THE TOXICOLOGY AND CARCINOGENESIS STUDIES OF FORMAMIDE (CAS NO. 75-12-7) IN F344/N RATS AND B6C3F1 MICE (GAVAGE STUDIES) NATIONAL TOXICOLOGY PROGRAM P.O. Box 12233 Research Triangle Park, NC 27709 July 2008 NTP TR 541 NIH Publication No. 08-5884 National Institutes of Health Public Health Service U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES FOREWORD The National Toxicology Program (NTP) is an interagency program within the Public Health Service (PHS) of the Department of Health and Human Services (HHS) and is headquartered at the National Institute of Environmental Health Sciences of the National Institutes of Health (NIEHS/NIH). Three agencies contribute resources to the program: NIEHS/NIH, the National Institute for Occupational Safety and Health of the Centers for Disease Control and Prevention (NIOSH/CDC), and the National Center for Toxicological Research of the Food and Drug Administration (NCTR/FDA). Established in 1978, the NTP is charged with coordinating toxicological testing activities, strengthening the science base in toxicology, developing and validating improved testing methods, and providing information about potentially toxic substances to health regulatory and research agencies, scientific and medical communities, and the public. The Technical Report series began in 1976 with carcinogenesis studies conducted by the National Cancer Institute. In 1981, this bioassay program was transferred to the NTP. The studies described in the Technical Report series are designed and conducted to characterize and evaluate the toxicologic potential, including carcinogenic activity, of selected substances in laboratory animals (usually two species, rats and mice). Substances selected for NTP toxicity and carcinogenicity studies are chosen primarily on the basis of human exposure, level of production, and chemical structure. -

United States Patent (19) 11 Patent Number: 4,572,296 Watkins W (45
United States Patent (19) 11 Patent Number: 4,572,296 Watkins w (45. Date of Patent: Feb. 25, 1986 (54) STEAM INJECTION METHOD Primary Examiner-George A. Suchfield Attorney, Agent, or Firm-Dean Sandford; Gregory F. (75) Inventor: David R. Watkins, Irvine, Calif. Wirzbicki; Robert A. Franks 73) Assignee: Union Oil Company of California, Los Angeles, Calif. (57) ABSTRACT 21 Appl. No.: 653,027 A method is disclosed for inhibiting silica dissolution during steam injection into a formation. In the method, 22 Filed: Sep. 20, 1984 there is added to the steam, or to feedwater used to 51) Int. Cl. .............................................. E21B 43/24 generate the steam, a mixture comprising: (52) ... 166/303; 166/902 (a) a compound selected from the group consisting of 58) Field of Search .................... 166/272,303, 244 C ammonium salts of inorganic acids, ammonium salts 56) References Cited of carboxylic acids, quaternary ammonium halides, amine or substituted amine hydrochlorides, and mix U.S. PATENT DOCUMENTS tures thereof; and 3,379,249 4/1968 Gilchrist et al. ................ 166/303 X (b) a compound selected from the group consisting of 3,822,749 7/1974. Thigpen, Jr. ........................ 166/303 ammonia, salts which decompose in steam to form 3,938,590 2/1976 Redford et al. 166/272 X 4, 156,463 5/1979 Hall ..................................... 166/272 acid neutralizers or buffers having alkaline pH values, 4,222,439 9/1980 Estes et al. ... 166/272 amides of carbamic acid or thiocarbamic acid and 4,223,731 9/1980 Estes et al. .. ... 166/272 derivatives of such amides, tertiary carboxylic acid 4,475,595 10/1984 Watkins et al. -

Evidence of Ammonium Salts in Comet 67P As Explanation for the Nitrogen Depletion in Cometary Comae
Evidence of ammonium salts in comet 67P as explanation for the nitrogen depletion in cometary comae Authors: K. Altwegg1*, H. Balsiger1, J.-J. Berthelier2, C. Briois3, M. Combi4, H. Cottin5, J. De Keyser6, F. Dhooghe6, B. Fiethe7, S. A. Fuselier8,9, T. I. Gombosi4, N. Hänni1, M. Rubin1, M. Schuhmann1, I. Schroeder1, T. Sémon1, S. Wampfler2 Affiliations: 1Physikalisches Institut, University of Bern, Sidlerstr. 5, CH-3012 Bern, Switzerland. 2Center for Space and Habitability, University of Bern, Sidlerstr. 5, CH-3012 Bern, Switzerland. 2LATMOS/IPSL-CNRS-UPMC-UVSQ, 4 Avenue de Neptune F-94100, Saint-Maur, France. 3Laboratoire de Physique et Chimie de l'Environnement et de l'Espace (LPC2E), UMR CNRS 7328 – Université d'Orléans, France 4Department of Climate and Space Sciences and Engineering, University of Michigan, 2455 Hayward, Ann Arbor, MI 48109, USA. 5 LISA, UMR CNRS 7583, Université Paris-Est-Créteil, Université de Paris, Institut Pierre Simon Laplace (IPSL), Créteil, France 6Royal Belgian Institute for Space Aeronomy, BIRA-IASB, Ringlaan 3, B-1180 Brussels, Belgium. 7Institute of Computer and Network Engineering (IDA), TU Braunschweig, Hans-Sommer- Straße 66, D-38106 Braunschweig, Germany. 8Space Science Directorate, Southwest Research Institute, 6220 Culebra Rd., San Antonio, TX 78228, USA. 9Department of Physics and Astronomy, The University of Texas at San Antonio, San Antonio, TX, 78249 Cometary comae are generally depleted in nitrogen. The main carriers for volatile nitrogen in comets are NH3 and HCN. It is known that ammonia readily combines with many acids like e.g. HCN, HNCO, HCOOH, etc. encountered in the interstellar medium as well as in + - cometary ice to form ammonium salts (NH4 X ) at low temperatures. -

Federal Register / Vol. 62, No. 116 / Tuesday, June 17, 1997 / Rules and Regulations
32974 Federal Register / Vol. 62, No. 116 / Tuesday, June 17, 1997 / Rules and Regulations ENVIRONMENTAL PROTECTION p.m. EST, toll-free, at 800±424±9346; standards for owners and operators of AGENCY 703±412±9810 from Government phones hazardous waste treatment, storage and or if in the Washington, DC local calling disposal facilitiesÐincluding standards 40 CFR Parts 261, 268, 271, and 302 area; or 800±553±7672 for the hearing for obtaining permitsÐare found at 40 impaired. For more detailed information CFR part 264. [EPA530±Z±97±FFF; FRL±5839±7] on specific aspects of the rulemaking, Hazardous wastes are also subject to RIN 2050±AD59 contact Caroline Gerwe by calling 703± land disposal restrictions under 40 CFR 308±3540 or by writing, to U.S. part 268. EPA's authorizations for state Hazardous Waste Management Environmental Protection Agency, hazardous waste programs are found at System; Carbamate Production, Office of Solid Waste, Hazardous Waste 40 CFR part 272. The requirements for Identification and Listing of Hazardous Identification Division, 401 M St., SW., obtaining these authorizations are found Waste; Land Disposal Restrictions; (Mailcode 5304W), Washington, DC at 40 CFR part 271. Authorization of State Hazardous 20460. In addition, hazardous wastes having Waste Programs; and CERCLA SUPPLEMENTARY INFORMATION: This rule the characteristics identified under, or Hazardous Substance Designation and is available on the Internet. Please listed pursuant to, RCRA section 3001 Reportable Quantities follow these instructions to access the (except when suspended by Congress) AGENCY: Environmental Protection rule electronically: From the World become hazardous substances under Agency (EPA). Wide Web (WWW), type http:// section 101(14)(C) of CERCLA, 42 U.S.C. -

An Access-Dictionary of Internationalist High Tech Latinate English
An Access-Dictionary of Internationalist High Tech Latinate English Excerpted from Word Power, Public Speaking Confidence, and Dictionary-Based Learning, Copyright © 2007 by Robert Oliphant, columnist, Education News Author of The Latin-Old English Glossary in British Museum MS 3376 (Mouton, 1966) and A Piano for Mrs. Cimino (Prentice Hall, 1980) INTRODUCTION Strictly speaking, this is simply a list of technical terms: 30,680 of them presented in an alphabetical sequence of 52 professional subject fields ranging from Aeronautics to Zoology. Practically considered, though, every item on the list can be quickly accessed in the Random House Webster’s Unabridged Dictionary (RHU), updated second edition of 2007, or in its CD – ROM WordGenius® version. So what’s here is actually an in-depth learning tool for mastering the basic vocabularies of what today can fairly be called American-Pronunciation Internationalist High Tech Latinate English. Dictionary authority. This list, by virtue of its dictionary link, has far more authority than a conventional professional-subject glossary, even the one offered online by the University of Maryland Medical Center. American dictionaries, after all, have always assigned their technical terms to professional experts in specific fields, identified those experts in print, and in effect held them responsible for the accuracy and comprehensiveness of each entry. Even more important, the entries themselves offer learners a complete sketch of each target word (headword). Memorization. For professionals, memorization is a basic career requirement. Any physician will tell you how much of it is called for in medical school and how hard it is, thanks to thousands of strange, exotic shapes like <myocardium> that have to be taken apart in the mind and reassembled like pieces of an unpronounceable jigsaw puzzle. -
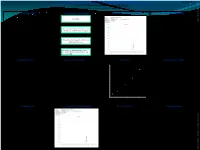
Quantification of Ethyl Carbamate in Tobacco
QUANTIFICATION OF ETHYL CARBAMATE IN TOBACCO POSTER-15 Dinesh.T.K., Sathya Gourishankar and Sharad.K.Mehta* ITC Ltd, ITC R&D Centre, Peenya, Bangalore – 560 058, INDIA. ABSTRACT SAMPLE PREPARATION SAMPLE CHROMATOGRAM REPRODUCIBILITY ETHYL 2.5g Sample CARBAMATE ANALYST TRIAL NO. SAMPLE (ng/g) 2012_TSRC15_Mehta.pdf A simple method is developed for quantification of Ethyl 1 Tobacco 371.0 carbamate in Tobacco. Ethyl carbamate in Tobacco is 2 Tobacco 368.7 extracted with water and is partitioned with Sonicate with 25ml water for 30 mins and 3 Tobacco 382.2 dichloromethane. The dichloromethane extract is Centrifuge at 10000 rpm for 10 minutes. 4 Tobacco 374.1 concentrated and injected in GC/MS-SIM mode. The 1 5 Tobacco 384.1 method has been validated using standard validation 6 Tobacco 389.0 protocols and there is excellent linearity over 7 Tobacco 368.3 concentration range from 100 to 4000 ng/g with Extract 10 ml of water extract with 30ml correlation coefficient of 0.9939. The recoveries are more dichloromethane twice. 8 Tobacco 367.1 than 90% for Ethyl carbamate with limit of quantification 9 Tobacco 364.5 100 ng/g and limit of detection 40ng/g respectively. carbamate Ethyl 2 10 Tobacco 372.2 Concentrate the dichloromethane extract STDEV 8.2 after passing through sodium sulphate and MEAN 374.1 inject in GC-MS. %RSD 2.2 INTRODUCTION GC-MS CONDITIONS LINEARITY RECOVERY STUDIES CALIBRATION FOR ETHYL CARBAMATE 400000 Ethyl carbamate (Urethane) is a genotoxic carcinogen found 350000 in fermented foods and beverages. Ethyl carbamate in y = 340.35x - 5869.2 SPIKED OBTAINED Tobacco is formed from Maleic hydrazide, which is a plant • Column : Zebron-ZB Wax plus 30mX0.25mmX0.25m.