QSAR Study of Aquatic Toxicity by Chemometrics Methods in the Framework of REACH Regulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds
Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Library Listing – 1,523 spectra Subset of Aldrich FT-IR Library related to organometallic, inorganic, boron and deueterium compounds. The Aldrich Material-Specific FT-IR Library collection represents a wide variety of the Aldrich Handbook of Fine Chemicals' most common chemicals divided by similar functional groups. These spectra were assembled from the Aldrich Collections of FT-IR Spectra Editions I or II, and the data has been carefully examined and processed by Thermo Fisher Scientific. Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Index Compound Name Index Compound Name 1066 ((R)-(+)-2,2'- 1193 (1,2- BIS(DIPHENYLPHOSPHINO)-1,1'- BIS(DIPHENYLPHOSPHINO)ETHAN BINAPH)(1,5-CYCLOOCTADIENE) E)TUNGSTEN TETRACARBONYL, 1068 ((R)-(+)-2,2'- 97% BIS(DIPHENYLPHOSPHINO)-1,1'- 1062 (1,3- BINAPHTHYL)PALLADIUM(II) CH BIS(DIPHENYLPHOSPHINO)PROPA 1067 ((S)-(-)-2,2'- NE)DICHLORONICKEL(II) BIS(DIPHENYLPHOSPHINO)-1,1'- 598 (1,3-DIOXAN-2- BINAPH)(1,5-CYCLOOCTADIENE) YLETHYNYL)TRIMETHYLSILANE, 1140 (+)-(S)-1-((R)-2- 96% (DIPHENYLPHOSPHINO)FERROCE 1063 (1,4- NYL)ETHYL METHYL ETHER, 98 BIS(DIPHENYLPHOSPHINO)BUTAN 1146 (+)-(S)-N,N-DIMETHYL-1-((R)-1',2- E)(1,5- BIS(DI- CYCLOOCTADIENE)RHODIUM(I) PHENYLPHOSPHINO)FERROCENY TET L)E 951 (1,5-CYCLOOCTADIENE)(2,4- 1142 (+)-(S)-N,N-DIMETHYL-1-((R)-2- PENTANEDIONATO)RHODIUM(I), (DIPHENYLPHOSPHINO)FERROCE 99% NYL)ETHYLAMIN 1033 (1,5- 407 (+)-3',5'-O-(1,1,3,3- CYCLOOCTADIENE)BIS(METHYLD TETRAISOPROPYL-1,3- IPHENYLPHOSPHINE)IRIDIUM(I) -

National Institute of Public Health and Environmental Protection Bilthoven the Netherlands
NATIONAL INSTITUTE OF PUBLIC HEALTH AND ENVIRONMENTAL PROTECTION BILTHOVEN THE NETHERLANDS Report no. 710401027 EXPLORATORY REPORT TIN AND TIN COMPOUNDS W. Slooff, P.F.H. Bont *, J.M. Hesse and J.A. Annema August 1993 Resources Planning Consultants BV, Delft This study has been carried out at the request and for the account of the Directorate- General for Environmental Protection, Direction Substances, Safety and Radiation /i Mailing list exploratory report Tin and tin compounds 1 Directoraat-Generaal voor Milieubeheer - Directie Stoffen, Veiligheid en Straling 2 Directeur-Generaal voor Milieubeheer 3- 4 Plv. Directeur-Generaal voor Milieubeheer 5 Dr. J. de Bruijn (DGM) 6 Dr. J.A. van Zorge (DGM) 7 Dr. A.G.J. Sedee (DGM) 8 Depot Nederlandse publicaties en Nederlandse bibliografie 9 Board of Directors of the National Institute of Public Health and Environmental Protection 10 Dr.ir. G. de Mik 11 Ir. A.H.M. Bresser 12 Mrs.Drs. A.G.A.C. Knaap 13 Prof Dr. H.A.M, de Kruijf 15-18 Authors 19-22 Laboratory for Ecotoxicology 23 Project and Report Registration 24 Report Administration 25 Head of Information and Public Relations 26-28 Library RIVM 29-38 Copies RPC 39-50 Spare copies RIVM CONTENTS SUMMARY iii 1. INTRODUCTION 1 2. ACTUAL STANDARDS AND GUIDELINES 2 3. APPLICATIONS. SOURCES AND EMISSIONS 5 3.1 PRODUCTION 5 3.2 APPLICATIONS 6 3.2.1 Metallic tin 6 3.2.2 Inorganic tin compounds 8 3.2.3 Organotin compounds 8 3.3 EMISSIONS AND WASTE STREAMS 10 3.3.1 Emissions 10 3.3.2 Waste streams 13 3.4 TRENDS 14 4 OCCURRENCE AND CONCENTRATIONS . -

M-Sw, SWU.T ISB3S, W.G.L V&H #U=- Lieponti All Rights Reserved
T o bctvtarae&l to the aoaus'n u ' mrgvsAR, •us i m ov l.-m-sw, SWU.t ISB3S, w.g.l v&h #u=- lieponti All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if materia! had to be removed, a note will indicate the deletion. Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 3WESTIGATI0NS OF SOME PHYSICAL PROPERTIES OF ORGATCIN COMPOUNDS A Thesis Submitted to the University of London for the Degree of Doctor of philosophy Uy GHAZI ABDUL WAHHAB DERWISH March 1959 (i) ABSTRACT The absorption spectra o f ten organotin compounds were measured in the in fra re d region 2 P 5-15 # 3 /t( 4000-650 ceT ) and in the u ltra vio le t region 200-400 The compounds used were: the homologous series P h^S nC l^ (n - 1,2,3,and 4), tetrabenzyltin, tribe n zyltin chloride, te tra -^ to ly ltin , t et rakis-£-chlorophenylt in * trie th y ltin phenoxide. and N s trie th y ltin phthalim ide» The results have been analysed and discussed in d e ta il and a tentative assignment of many of the normal vib ratio n a l frequencies carried out. -

Molecular Summary Tables
Summary Tables of Organic, Silicon, Boron, Aluminum and Organometallic Molecules and Exemplary Results on Condensed Matter Physics Tables summarizing the results of the calculated experimental parameters of 800 exemplary solved molecules follow. The closed-form derivations of these molecules can be found in The Grand Theory of Classical Physics posted at http://www.blacklightpower.com/theory/bookdownload.shtml Chapters 15–17, as well as Silicon in Chapter 20, Boron in Chapter 22, and Aluminum and Organometallics in Chapter 23. Condensed matter physics based on first principles with analytical solutions of (i) of the geometrical parameters and energies of the hydrogen bond of H2O in the ice and steam phases, and of H2O and NH3; (ii) analytical solutions of the geometrical parameters and interplane van der Waals cohesive energy of graphite; (iii) analytical solutions of the geometrical parameters and interatomic van der Waals cohesive energy of liquid helium and solid neon, argon, krypton, and xenon are given in Chapter 16. 1 SUMMARY TABLES OF ORGANIC, SILICON, BORON, ORGANOMETALLIC, AND COORDNINATE MOLECULES The results of the determination of the total bond energies with the experimental values are given in the following tables for a large array of functional groups and molecules per class for which the experimental data was available. Here, the total bond energies of exemplary organic, silicon, boron, organometallic, and coordinate molecules whose designation is based on the main functional group were calculated using the functional group composition and the corresponding energies derived previously [1] and compared to the experimental values. References for the experimental values are mainly from Ref. -

Environmental Health Criteria 15 TIN and ORGANOTIN COMPOUNDS A
Environmental Health Criteria 15 TIN AND ORGANOTIN COMPOUNDS A Preliminary Review Please note that the layout and pagination of this web version are not identical with the printed version. Tin and organotin compounds (EHC 15, 1980) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 15 TIN AND ORGANOTIN COMPOUNDS A Preliminary Review This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of either the World Health Organization or the United Nations Environment Programme. Published under the joint sponsorship of the United Nations Environment Programme and the World Health Organization World Health Organization Geneva, 1980 ISBN 92 4 154075 3 (c) World Health Organization 1980 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. For rights of reproduction or translation of WHO publications, in part or in toto, application should be made to the Office of Publications, World Health Organization, Geneva, Switzerland. The World Health Organization welcomes such applications. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturers' products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. -

Total Bond Energies of Exact Classical Solutions of Molecules Generated by Millsian 1.0 Compared to Those Computed Using Modern 3-21G and 6-31G* Basis Sets
Total Bond Energies of Exact Classical Solutions of Molecules Generated by Millsian 1.0 Compared to Those Computed Using Modern 3-21G and 6-31G* Basis Sets R. L. Mills, B. Holverstott, B. Good, N. Hogle, A. Makwana Millsian, Inc., 493 Old Trenton Road, Cranbury, NJ 08512, www.millsian.com ABSTRACT Mills [1-12] solved the structure of the bound electron using classical laws and subsequently developed a unification theory based on those laws called the Grand Unified Theory of Classical Physics (GUTCP) with results that match observations for the basic phenomena of physics and chemistry from the scale of the quarks to cosmos. Millsian 1.0 is a program comprising molecular modeling applications of GUTCP, solving atomic and molecular structures based on applying the classical laws of physics, (Newton’s and Maxwell’s Laws) to the atomic scale. The functional groups of all major classes of chemical bonding including those involved in most organic molecules have been solved exactly in closed-form solutions. By using these functional groups as building blocks, or independent units, a potentially infinite number of molecules can be solved. As a result, Millsian software can visualize the exact three-dimensional structure and calculate physical characteristics of almost any molecule of any length and complexity. Even complex proteins and DNA (the molecules that encode genetic information) can be solved in real-time interactively on a personal computer. By contrast, previous software based on traditional quantum methods must resort to approximations and run on powerful computers for even the simplest systems. The energies of exact classical solutions of molecules generated by Millsian 1.0 and those from a modern quantum mechanics-based program, Spartan’s pre- computed database using 3-21G and 6-31G* basis sets at the Hartree-Fock level of theory, were compared to experimental values. -

A Sheffield Hallam University Thesis
The chemistry of some heteroaryltin compounds. DERBYSHIRE, Diana Jane. Available from the Sheffield Hallam University Research Archive (SHURA) at: http://shura.shu.ac.uk/19555/ A Sheffield Hallam University thesis This thesis is protected by copyright which belongs to the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Please visit http://shura.shu.ac.uk/19555/ and http://shura.shu.ac.uk/information.html for further details about copyright and re-use permissions. I P G I A III Sheffield City Polytechnic Library REFERENCE ONLY I ProQuest Number: 10694436 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10694436 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 THE CHEMISTRY OF SOME HETEROARYLTIN COMPOUNDS by DIANA JANE DERBYSHIRE A Thesis submitted to the Council -

Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds
Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Library Listing – 1,521 spectra Subset of Aldrich FT-IR Library related to organometallic, inorganic, boron and deuterium compounds. The Aldrich Material-Specific FT-IR Library collection represents a wide variety of the Aldrich Handbook of Fine Chemicals' most common chemicals divided by similar functional groups. These spectra were assembled from the Aldrich Collection of FT-IR Spectra and the data has been carefully examined and processed by Thermo. The molecular formula, CAS (Chemical Abstracts Services) registry number, when known, and the location number of the printed spectrum in The Aldrich Library of FT-IR Spectra are available. Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Index Compound Name Index Compound Name 1066 ((R)-(+)-2,2'- 1062 (1,3- BIS(DIPHENYLPHOSPHINO)-1,1'- BIS(DIPHENYLPHOSPHINO)PROPA BINAPH)(1,5-CYCLOOCTADIENE) NE)DICHLORONICKEL(II) 1068 ((R)-(+)-2,2'- 598 (1,3-DIOXAN-2- BIS(DIPHENYLPHOSPHINO)-1,1'- YLETHYNYL)TRIMETHYLSILANE, BINAPHTHYL)PALLADIUM(II) CH 96% 1067 ((S)-(-)-2,2'- 1063 (1,4- BIS(DIPHENYLPHOSPHINO)-1,1'- BIS(DIPHENYLPHOSPHINO)BUTAN BINAPH)(1,5-CYCLOOCTADIENE) E)(1,5- 1140 (+)-(S)-1-((R)-2- CYCLOOCTADIENE)RHODIUM(I) (DIPHENYLPHOSPHINO)FERROCE TET NYL)ETHYL METHYL ETHER, 98 951 (1,5-CYCLOOCTADIENE)(2,4- 1146 (+)-(S)-N,N-DIMETHYL-1-((R)-1',2- PENTANEDIONATO)RHODIUM(I), BIS(DI- 99% PHENYLPHOSPHINO)FERROCENY 1033 (1,5- L)E CYCLOOCTADIENE)BIS(METHYLD 1142 (+)-(S)-N,N-DIMETHYL-1-((R)-2- IPHENYLPHOSPHINE)IRIDIUM(I) -

Toxicological Profile for Tin and Tin Compounds
TOXICOLOGICAL PROFILE FOR TIN AND TIN COMPOUNDS U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry August 2005 TIN AND TIN COMPOUNDS ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. TIN AND TIN COMPOUNDS iii UPDATE STATEMENT A Toxicological Profile for Tin and Tin Compounds, Draft for Public Comment was released in September 2003. This edition supersedes any previously released draft or final profile. Toxicological profiles are revised and republished as necessary. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE Mailstop F-32 Atlanta, Georgia 30333 TIN AND TIN COMPOUNDS vi *Legislative Background The toxicological profiles are developed in response to the Superfund Amendments and Reauthorization Act (SARA) of 1986 (Public law 99-499) which amended the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund). This public law directed ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. The availability of the revised priority list of 275 hazardous substances was announced in the Federal Register on November 17, 1997 (62 FR 61332). For prior versions of the list of substances, see Federal Register notices dated April 29, 1996 (61 FR 18744); April 17, 1987 (52 FR 12866); October 20, 1988 (53 FR 41280); October 26, 1989 (54 FR 43619); October 17, 1990 (55 FR 42067); October 17, 1991 (56 FR 52166); October 28, 1992 (57 FR 48801); and February 28, 1994 (59 FR 9486). -

United States Patent Office Patented Dec
3,629,215 United States Patent Office Patented Dec. 21, 1971 2 3,629,215 wherein R and R2 are respectively hydrogen atom, halo 1:1. ALTERNATING COPOLYMERS OF SUBST gen atoms, hydrocarbon radicals having 1 to 20 carbon TUTED CONUGATED WNY COMPOUNDS atoms, halogen-containing hydrocarbon radicals having AND OLEFNC COMPOUNDS AND PROCESS 1 to 20 carbon atoms, or substituted radicals thereof, FOR PRODUCING THE SAME and B) a substituted conjugated compound having the Kohei Nakaguchi, Osaka, Shohachi Kawasumi, Kobe, formula: Masaaki Hirooka, Ibaraki-shi, Hiroshi Yabuuchi, Takat suki-shi, and Hiroyoshi Takao, Ashiya-shi, Japan, as RIE ises to Sumitomo Chemical Company, Ltd., Osaka, RICH-C-Q apan No Drawing. Continuation of application Ser. No. 10 wherein R and R are respectively hydrogen or halogen 567,353, July 25, 1966. This application Oct. 31, atoms or hydrocarbon radicals having 1 to 20 carbon 1969, Ser. No. 871,781 atoms, halogen-containing hydrocarbon radicals having Claims priority, application Japan, July 30, 1965, 1 to 20 carbon atoms, substituted hydrocarbon radicals 40/46,495 thereof, either of RI and RI is hydrogen atom but both Int. C. C08f3/40, 3/62, 3/74 5 of them are not hydrogen at the same time; and Q is a U.S. C. 260-85.5 HC 41 Claims nitrile or ABSTRACT OF THE DISCLOSURE An alternating copolymer consists of AJ a terminally 20 group, wherein Y is Z2H, Z2R, Z2Me, MR'R'', R, halo unsaturated olefinic compound, for example, an ox-olefin, gen atoms or hydrogen atom; in which Z and Z are an isoolefin, or a halogenated olefinic compound -
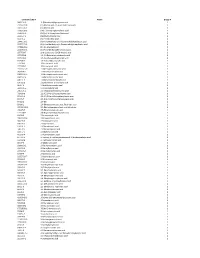
Chemical Classification List
Identifier/CAS # Name Group # 5683-31-8 3-(Trimethylsilyl)propynoic acid 1 21293-29-8 (+)-Abscisic acid, (2, 4-pentadienoic acid) 1 14375-45-2 (±)-Abscisic acid 1 35963-20-3 (1R)-(-)-10-Camphorsulfonic acid 1 3144-16-9 (1S)-(+)-10-Camphorsulfonic acid 1 2444-37-3 (Methylthio)acetic acid 1 611-71-2 (R)-(−)-Mandelic acid 1 20445-31-2 (R)-(+)-α-Methoxy-α-trifluoromethylphenylacetic acid 1 17257-71-5 (S)-(−)-α-Methoxy-α-(trifluoromethyl)phenylacetic acid 1 17199-29-0 (S )-(+)-Mandelic acid 1 26164-26-1 (S)-(+)-α-Methoxyphenylacetic acid 1 1077-28-7 ±)-α-Lipoic acid, DL-6,8-thioctic acid 1 1703-58-8 1,2,3,4-Butanetetracarboxylic acid 1 3971-31-1 1,3-Cyclohexanedicarboxylic acid 1 542-05-2 1,3-Acetonedicarboxylic acid 1 112-38-9 10-Undecenoic acid 1 2777-65-3 10-Undecynoic acid 1 71310-21-9 11-Mercaptoundecanoic acid 1 2834-05-1 11-Bromoundecanoic acid 1 69839-68-5 16-Mercaptohexadecanoic acid 1 4942-47-6 1-Adamantaneacetic acid 1 828-51-3 1-Adamantanecarboxylic acid 1 636-82-8 1-Cyclohexene-1-carboxylic acid 1 86-87-3 1-Naphthaleneacetic acid 1 3443-45-6 1-Pyrenebutyric acid 1 2062-25-1 2-(Trifluoromethyl)cinnamic acid 1 719-60-8 2,3,4,5,6-Pentafluorocinnamic acid 1 653-21-4 2,3,4,5,6-Pentafluorophenylacetic acid 1 94-75-7 2,4-D (2,4-Dichlorophenoxyacetic acid) 1 94-82-6 2,4-DB 1 89-86-1 2,4-Dihydroxybenzoic acid, Resorcylic acid 1 207234-00-2 2,4-Dinitrobenzenesulfonic acid dihydrate 1 490-79-9 2,5-Dihydroxybenzoic acid 1 1141-38-4 2,6-Naphthalenedicarboxylic acid 1 80-58-0 2-Bromobutyric acid 1 70610-87-6 2-Bromooctanoic acid 1 584-93-0