Human Mutation Published by Wiley Periodicals, Inc
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Defect in NIPAL4 Is Associated with Autosomal Recessive Congenital Ichthyosis in American Bulldogs
RESEARCH ARTICLE A Defect in NIPAL4 Is Associated with Autosomal Recessive Congenital Ichthyosis in American Bulldogs Margret L. Casal1*, Ping Wang1, Elizabeth A. Mauldin2, Gloria Lin1, Paula S. Henthorn1 1 Section of Medical Genetics, Department of Clinical Studies, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America, 2 Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America a1111111111 * [email protected] a1111111111 a1111111111 a1111111111 a1111111111 Abstract Autosomal recessive congenital ichthyosis in the American bulldog is characterized by gen- eralized scaling and erythema with adherent scale on the glabrous skin. We had previously linked this disorder to NIPAL4, which encodes the protein ichthyin. Sequencing of NIPAL4 OPEN ACCESS revealed a homozygous single base deletion (CanFam3.1 canine reference genome Citation: Casal ML, Wang P, Mauldin EA, Lin G, sequence NC_06586.3 g.52737379del), the 157th base (cytosine) in exon 6 of NIPAL4 as Henthorn PS (2017) A Defect in NIPAL4 Is the most likely causative variant in affected dogs. This frameshift deletion results in a prema- Associated with Autosomal Recessive Congenital ture stop codon producing a truncated and defective NIPAL4 (ichthyin) protein of 248 amino Ichthyosis in American Bulldogs. PLoS ONE 12(1): e0170708. doi:10.1371/journal.pone.0170708 acids instead of the wild-type length of 404. Obligate carriers were confirmed to be heterozy- gous for this variant, and 150 clinically non-affected dogs of other breeds were homozygous Editor: Claire Wade, University of Sydney Faculty of Veterinary Science, AUSTRALIA for the wild-type gene. Among 800 American bulldogs tested, 34% of clinically healthy dogs were discovered to be heterozygous for the defective allele. -

Transcriptome Profiling and Differential Gene Expression In
G C A T T A C G G C A T genes Article Transcriptome Profiling and Differential Gene Expression in Canine Microdissected Anagen and Telogen Hair Follicles and Interfollicular Epidermis Dominique J. Wiener 1,* ,Kátia R. Groch 1 , Magdalena A.T. Brunner 2,3, Tosso Leeb 2,3 , Vidhya Jagannathan 2 and Monika M. Welle 3,4 1 Department of Veterinary Pathobiology, College of Veterinary Medicine & Biomedical Science, Texas A&M University, College Station, TX 77843, USA; [email protected] 2 Institute of Genetics, Vetsuisse Faculty, University of Bern, 3012 Bern, Switzerland; [email protected] (M.A.T.B.); [email protected] (T.L.); [email protected] (V.J.) 3 Dermfocus, Vetsuisse Faculty, University Hospital of Bern, 3010 Bern, Switzerland; [email protected] 4 Institute of Animal Pathology, Vetsuisse Faculty, University of Bern, 3012 Bern, Switzerland * Correspondence: [email protected]; Tel.: +1-979-862-1568 Received: 30 June 2020; Accepted: 3 August 2020; Published: 4 August 2020 Abstract: The transcriptome profile and differential gene expression in telogen and late anagen microdissected hair follicles and the interfollicular epidermis of healthy dogs was investigated by using RNAseq. The genes with the highest expression levels in each group were identified and genes known from studies in other species to be associated with structure and function of hair follicles and epidermis were evaluated. Transcriptome profiling revealed that late anagen follicles expressed mainly keratins and telogen follicles expressed GSN and KRT15. The interfollicular epidermis expressed predominately genes encoding for proteins associated with differentiation. All sample groups express genes encoding for proteins involved in cellular growth and signal transduction. -

Multilocus Disease-Causing Genomic Variations for Mendelian Disorders: Role of Systematic Phenotyping and Implications on Genetic Counselling
www.nature.com/ejhg ARTICLE OPEN Multilocus disease-causing genomic variations for Mendelian disorders: role of systematic phenotyping and implications on genetic counselling Dhanya Lakshmi Narayanan 1, Divya Udyawar1, Parneet Kaur1, Suvasini Sharma2, Narayanaswamy Suresh2, Sheela Nampoothiri 3, 1 1 1 1 1 1 Michelle C. do Rosario , Puneeth H. Somashekar , Lakshmi Priya Rao , Neethukrishna Kausthubham✉ , Purvi Majethia , Shruti Pande , Y. Ramesh Bhat4, Aroor Shrikiran4, Stephanie Bielas5, Katta Mohan Girisha 1 and Anju Shukla 1 © The Author(s) 2021 Multilocus disease-causing genomic variations (MGVs) and multiple genetic diagnoses (MGDs) are increasingly being recognised in individuals and families with Mendelian disorders. This can be mainly attributed to the widespread use of genomic tests for the evaluation of these disorders. We conducted a retrospective study of families evaluated over the last 6 years at our centre to identify families with MGVs and MGDs. MGVs were observed in fourteen families. We observed five different consequences: (i) individuals with MGVs presenting as blended phenotypes (ii) individuals with MGVs presenting with distinct phenotypes (iii) individuals with MGVs with age-dependent penetrance (iv) individuals with MGVs with one phenotype obscured by another more predominant phenotype (v) two distinct phenotypes in different individuals in families with MGVs. Consanguinity was present in eight (8/14, 57.1%) of them. Thirteen families had two Mendelian disorders and one had three Mendelian disorders. The risk of recurrence of one or more conditions in these families ranged from 25% to 75%. Our findings underline the importance of the role of a clinical geneticist in systematic phenotyping, challenges in genetic counselling and risk estimation in families with MGVs and MGDs, especially in highly inbred populations. -

Gnomad Lof Supplement
1 gnomAD supplement gnomAD supplement 1 Data processing 4 Alignment and read processing 4 Variant Calling 4 Coverage information 5 Data processing 5 Sample QC 7 Hard filters 7 Supplementary Table 1 | Sample counts before and after hard and release filters 8 Supplementary Table 2 | Counts by data type and hard filter 9 Platform imputation for exomes 9 Supplementary Table 3 | Exome platform assignments 10 Supplementary Table 4 | Confusion matrix for exome samples with Known platform labels 11 Relatedness filters 11 Supplementary Table 5 | Pair counts by degree of relatedness 12 Supplementary Table 6 | Sample counts by relatedness status 13 Population and subpopulation inference 13 Supplementary Figure 1 | Continental ancestry principal components. 14 Supplementary Table 7 | Population and subpopulation counts 16 Population- and platform-specific filters 16 Supplementary Table 8 | Summary of outliers per population and platform grouping 17 Finalizing samples in the gnomAD v2.1 release 18 Supplementary Table 9 | Sample counts by filtering stage 18 Supplementary Table 10 | Sample counts for genomes and exomes in gnomAD subsets 19 Variant QC 20 Hard filters 20 Random Forest model 20 Features 21 Supplementary Table 11 | Features used in final random forest model 21 Training 22 Supplementary Table 12 | Random forest training examples 22 Evaluation and threshold selection 22 Final variant counts 24 Supplementary Table 13 | Variant counts by filtering status 25 Comparison of whole-exome and whole-genome coverage in coding regions 25 Variant annotation 30 Frequency and context annotation 30 2 Functional annotation 31 Supplementary Table 14 | Variants observed by category in 125,748 exomes 32 Supplementary Figure 5 | Percent observed by methylation. -

Mining Patterns in Genomic and Clinical Cancer Data to Characterize Novel Driver Genes
Mining patterns in genomic and clinical cancer data to characterize novel driver genes Rachel D. Melamed Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015 ©2015 Rachel D. Melamed All rights reserved ABSTRACT Mining patterns in genomic and clinical cancer data to characterize novel driver genes Rachel D. Melamed Cancer research, like many areas of science, is adapting to a new era characterized by increasing quantity, quality, and diversity of observational data. An example of the advances, and the resulting challenges, is represented by The Cancer Genome Atlas, an enormous public effort that has provided genomic profiles of hundreds of tumors of each of the most common solid cancer types. Alongside this resource is a host of other data and knowledge, including gene interaction databases, Mendelian disease causal variants, and electronic health records spanning many millions of patients. Thus, a current challenge is how best to integrate these data to discover mechanisms of oncogenesis and cancer progression. Ultimately, this could enable genomics- based prediction of an individual patient’s outcome and targeted therapies, a goal termed precision medicine. In this thesis, I develop novel approaches that examine patterns in populations of cancer patients to identify key genetic changes and suggest likely roles of these driver genes in the diseases. In the first section I show how genomics can lead to the identification of driver alterations in melanoma. The most recurrent genetic mutations are often in important cancer driver genes: in a newly sequenced melanoma cohort, recurrent inactivating mutations point to an exciting new melanoma candidate tumor suppressor, FBXW7, with therapeutic implications. -

The 15Q11.2 BP1-BP2 Microdeletion
International Journal of Molecular Sciences Article The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders Syed K. Rafi * and Merlin G. Butler * Departments of Psychiatry & Behavioral Sciences and Pediatrics, University of Kansas Medical Center, Kansas City, KS 66160, USA * Correspondence: rafi[email protected] (S.K.R.); [email protected] (M.G.B.); Tel.: +816-787-4366 (S.K.R.); +913-588-1800 (M.G.B.) Received: 10 February 2020; Accepted: 29 April 2020; Published: 6 May 2020 Abstract: The 15q11.2 BP1-BP2 microdeletion (Burnside–Butler) syndrome is emerging as the most frequent pathogenic copy number variation (CNV) in humans associated with neurodevelopmental disorders with changes in brain morphology, behavior, and cognition. In this study, we explored functions and interactions of the four protein-coding genes in this region, namely NIPA1, NIPA2, CYFIP1, and TUBGCP5, and elucidate their role, in solo and in concert, in the causation of neurodevelopmental disorders. First, we investigated the STRING protein-protein interactions encompassing all four genes and ascertained their predicted Gene Ontology (GO) functions, such as biological processes involved in their interactions, pathways and molecular functions. These include magnesium ion transport molecular function, regulation of axonogenesis and axon extension, regulation and production of bone morphogenetic protein and regulation of cellular growth and development. We gathered a list of significantly associated cardinal maladies for each gene from searchable genomic disease websites, namely MalaCards.org: HGMD, OMIM, ClinVar, GTR, Orphanet, DISEASES, Novoseek, and GeneCards.org. Through tabulations of such disease data, we ascertained the cardinal disease association of each gene, as well as their expanded putative disease associations. -

Pdf) and the Common Can Be Grouped Into a Module of Associated Terms International Association for the Study of Pain (IASP)
MOLECULAR MEDICINE REPORTS 19: 1529-1542, 2019 Systematic screening and identification of novel psoriasis‑specific genes from the transcriptome of psoriasis‑like keratinocytes ZHEN WANG1*, HUAPING ZHENG1*, HONG ZHOU1*, NONGYU HUANG1, XIAOQIONG WEI1, XIAO LIU1, XIU TENG1, ZHONGLAN HU1, JUN ZHANG1, XIKUN ZHOU1, WEI LI2 and JIONG LI1 1Department of Biotherapy and Cancer Center; 2Department of Dermatovenereology, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, P.R. China Received March 28, 2018; Accepted November 5, 2018 DOI: 10.3892/mmr.2018.9782 Abstract. Psoriasis is a chronic inflammatory skin disease. Introduction Keratinocytes (KCs), as skin‑specific cells, serve an important role in the immunopathogenesis of psoriasis. In the present Skin functions as an important natural barrier between an study, transcriptome data derived from psoriasis‑like KCs organism and its external environment, thus it has a unique were used together with the reported transcriptome data from biological structure and specific immune functions (1). Skin is the skin/epidermis of patient with psoriasis, excluding known composed of two distinct regions, the epidermis and dermis. psoriasis‑associated genes that have been well described in the The predominant cell type of the epidermis is keratinocytes previous studies according to GeneCards database, to screen (KCs), whereas the dermis contains cells of the immune system, for novel psoriasis‑associated genes. According to the human including dendritic cells (DCs), T helper cells, γδT cells, natural expressed sequence tag of UniGene dataset, six genes that are killer T cells, macrophages and fibroblasts (2). A skin immune located near psoriasis‑associated loci were highly expressed system imbalance can cause several immune‑mediated skin in skin. -

High-Throughput Analysis of WNT Signaling Pathway in Osteoblasts
UNIVERSITY OF CALIFORNIA, MERCED High-throughput Analysis of WNT Signaling Pathway in Osteoblasts A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Quantitative and Systems Biology by Aimy Sebastian Committee in charge: Dr. Suzanne S. Sindi, Chair Dr. David H. Ardell Dr. Gabriela G. Loots Dr. Jennifer O. Manilay 2016 Copyright Aimy Sebastian, 2016 All rights reserved The Dissertation of Aimy Sebastian is approved, and it is acceptable in quality and form for publication on microfilm and electronically: Gabriela G. Loots Jennifer O. Manilay David H. Ardell Suzanne S. Sindi, Chair Date University of California, Merced 2016 iii Table of Contents Signature Page ..................................................................................................................... iii Table of Contents ................................................................................................................. iv List of Figures ...................................................................................................................... vi List of Tables ..................................................................................................................... viii Acknowledgements .............................................................................................................. ix VITA ..................................................................................................................................... x Abstract ............................................................................................................................. -
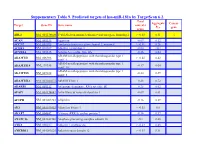
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Identification of a Novel Missense Mutation in NIPAL4 Gene: First 3D Model Construction Predicted Its Pathogenicity
Received: 27 October 2019 | Revised: 4 December 2019 | Accepted: 10 December 2019 DOI: 10.1002/mgg3.1104 ORIGINAL ARTICLE Identification of a novel missense mutation in NIPAL4 gene: First 3D model construction predicted its pathogenicity Sahar Laadhar1 | Riadh Ben Mansour2,3 | Slaheddine Marrakchi4 | Nabil Miled5 | Mariem Ennouri1 | Judith Fischer6 | Mohamed Ali Kaddechi7 | Hamida Turki4 | Faiza Fakhfakh1 1Faculty of Sciencs of Sfax, Laboratory of Molecular and Functional Genetics, Sfax, Abstract Tunisia Background: The NIPAL4 gene is described to be implicated of Congenital 2Laboratory of Food Analysis Valorization Ichthyosiform Erythroderma (CIE). It encodes a magnesium transporter membrane- and Security, Research Group associated protein, hypothetically involved in epidermal lipid processing and in la- "Biotechnology and pathologies", National School of Engineer of Sfax, Sfax, Tunisia mellar body formation. The aim of this work is to investigate the causative mutation 3Faculty of Sciences of Gafsa, Department in a consanguineous Tunisian family with a clinical feature of CIE with a yellowish of Life Sciences, Gafsa, Tunisia severe palmoplantar keratoderma. 4 Dermatology Department, HediChaker Methods: Four patients were dignosed with CIE. The blood samples were collected Hospital, Sfax, Tunisia from patients and all members of their nuclear family for mutation analysis. The 5Faculty of Sciences, Department of Biological Sciences, University of Jeddah, novel mutation of NIPAL4 gene was analysed with several software tools to predict Jeddah, KSA its pathogenicity. Then, the secondary structure and the 3D model of ichthyn was 6 Institute of Human Genetics, Medical generated in silico. Center-University of Freiburg, Faculty of NIPAL4 Medicine, University of Freiburg, Freiburg, Results: The sequencing analysis of the gene in patients revealed a novel Germany homozygous missense mutation c.534A>C (p.E178D) in the exon 4. -

Glomerular Expression Pattern of Long Non-Coding Rnas in the Type 2
www.nature.com/scientificreports OPEN Glomerular expression pattern of long non-coding RNAs in the type 2 diabetes mellitus BTBR mouse Received: 13 August 2018 Accepted: 11 June 2019 model Published: xx xx xxxx Simone Reichelt-Wurm 1, Tobias Wirtz1, Dominik Chittka1, Maja Lindenmeyer2,3, Robert M. Reichelt4, Sebastian Beck1, Panagiotis Politis5, Aristidis Charonis6, Markus Kretz7, Tobias B. Huber3, Shuya Liu3, Bernhard Banas 1 & Miriam C. Banas1 The prevalence of type 2 diabetes mellitus (T2DM) and by association diabetic nephropathy (DN) will continuously increase in the next decades. Nevertheless, the underlying molecular mechanisms are largely unknown and studies on the role of new actors like long non-coding RNAs (lncRNAs) barely exist. In the present study, the inherently insulin-resistant mouse strain “black and tan, brachyuric” (BTBR) served as T2DM model. While wild-type mice do not exhibit pathological changes, leptin- defcient diabetic animals develop a severe T2DM accompanied by a DN, which closely resembles the human phenotype. We analyzed the glomerular expression of lncRNAs from wild-type and diabetic BTBR mice (four, eight, 16, and 24 weeks) applying the “GeneChip Mouse Whole Transcriptome 1.0 ST” array. This microarray covered more lncRNA gene loci than any other array before. Over the observed time, our data revealed diferential expression patterns of 1746 lncRNAs, which markedly difered from mRNAs. We identifed protein-coding and non-coding genes, that were not only co-located but also co- expressed, indicating a potentially cis-acting function of these lncRNAs. In vitro-experiments strongly suggested a cell-specifc expression of these lncRNA-mRNA-pairs. Additionally, protein-coding genes, being associated with signifcantly regulated lncRNAs, were enriched in various biological processes and pathways, that were strongly linked to diabetes.