Chem Soc Rev 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amide Bond Formation and Peptide Coupling
Tetrahedron 61 (2005) 10827–10852 Tetrahedron report number 740 Amide bond formation and peptide coupling Christian A. G. N. Montalbetti* and Virginie Falque Evotec, 112 Milton Park, Abingdon OX14 4SD, UK Received 2 August 2005 Available online 19 September 2005 Contents 1. Introduction ................................................................. 10828 2. Amide bond formation: methods and strategies ....................................... 10828 2.1. Acyl halides . .......................................................... 10829 2.1.1. Acyl chlorides .................................................... 10829 2.1.1.1. Acyl chloride formation ...................................... 10829 2.1.1.2. Coupling reactions with acyl chlorides ........................... 10831 2.1.1.3. Limitations of acyl chlorides .................................. 10831 2.1.2. Acyl fluorides .................................................... 10831 2.1.3. Acyl bromides .................................................... 10832 2.2. Acyl azides . .......................................................... 10832 2.3. Acylimidazoles using CDI ................................................. 10833 2.4. Anhydrides . .......................................................... 10834 2.4.1. Symmetric anhydrides .............................................. 10834 2.4.2. Mixed anhydrides .................................................. 10834 2.4.2.1. Mixed carboxylic anhydrides .................................. 10834 2.4.2.2. Mixed carbonic anhydrides ................................... -

Reduction of Organic Functional Groups Using Hypophosphites Rim Mouselmani
Reduction of Organic Functional Groups Using Hypophosphites Rim Mouselmani To cite this version: Rim Mouselmani. Reduction of Organic Functional Groups Using Hypophosphites. Other. Univer- sité de Lyon; École Doctorale des Sciences et de Technologie (Beyrouth), 2018. English. NNT : 2018LYSE1241. tel-02147583v2 HAL Id: tel-02147583 https://tel.archives-ouvertes.fr/tel-02147583v2 Submitted on 5 Jun 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THESE de DOCTORAT DE L’UNIVERSITE DE LYON EN COTUTELLE AVEC L'UNIVERSITÉ LIBANAISE opérée au sein de l’Université Claude Bernard Lyon 1 École Doctorale de Chimie-École Doctorale des Sciences et Technologies Discipline : Chimie Soutenue publiquement le 07/11/2018, par Rim MOUSELMANI Reduction of Organic Functional Groups Using Hypophosphites Devant le jury composé de Mme. Micheline DRAYE Université Savoie Mont Blanc Rapporteure M. Mohammad ELDAKDOUKI Université Arabe de Beyrouth Rapporteur Mme. Emmanuelle SCHULZ Université Paris 11 examinatrice M. Abderrahmane AMGOUNE Université Lyon 1 Président M. Mahmoud FARAJ Université Internationale Libanaise examinateur Mme. Estelle MÉTAY Université Lyon 1 Directrice de thèse M. Ali HACHEM Université Libanaise Directeur de thèse M. Marc LEMAIRE Université Lyon 1 Membre invité M. -

Amide, and Paratoluenesulfonamide on the Amide of Silver,On the Imides
68 CHEMISTRY: E. C. FRANKLIN METALLIC SALTS OF AMMONO ACIDS By Edward C. Franklin DEPARTMENT OF CHEMISTRY, STANFORD UNIVERSITY Presented to the Academy, January 9. 1915 The Action of Liquid-Ammonia Solutions of Ammono Acids on Metallic Amides, Imides, and Nitrides. The acid amides and imides, and the metallic derivatives of the acid amides and imides are the acds, bases, and salts respectively of an ammonia system of acids, bases, and salts.1 Guided by the relationships implied in the above statement Franklin and Stafford were able to prepare potassium derivatives of a considerable number of acid amides by the action of potassium amide on certain acid amides in solution in liquid ammonia. That is to say, an ammono base, potassium amide, was found to react with ammono acids in liquid ammonia to form ammono salts just as the aquo base, potassium hydrox- ide, acts upon aquo acids in water solution to form aquo salts. Choos- ing, for example, benzamide and benzoic acid as representative acids of the two systems, the analogous reactions taking place respectively in liquid ammonia and water are represented by the equations: CH6CONH2+KNH2 = C6H5CONHK + NHs. CseHCONH2 + 2KNH2 = CIHsCONK2 + 2NH3. CH6tCOOH + KOH = CIH6COOK + H2O. The ammono acid, since it is dibasic, reacts with either one or two molecules of potassium amide to form an acid and a neutral salt. Having thus demonstrated the possibility of preparing ammono salts of potassium by the interaction of potassium amide and acid amides in liquid ammonia solution, it was further found that ammono salts of the heavy metals may be prepared by the action of liquid ammonia solutions of ammono acids on insoluble metallic amides, imides, and nitrides-that is, by reactions which are analogous to the formation of aquo salts in water by the action of potassium hydroxide on insoluble metallic hydroxides and oxides. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Activation of Pyridinium Salts for Electrophilic Acylation: a Method for Conversion of Pyridines Into 3-Acylpyridines
Chemistry of Heterocyclic Compounds, Vol. 40, No. 6, 2004 ACTIVATION OF PYRIDINIUM SALTS FOR ELECTROPHILIC ACYLATION: A METHOD FOR CONVERSION OF PYRIDINES INTO 3-ACYLPYRIDINES A. Klapars1 and E. Vedejs2 Cyanide adducts of N-MOM pyridinium salts react with strong acylating reagents to provide 3-acyl-4- cyano-1,4-dihydropyridines that can be aromatized to 3-acylpyridines using ZnCl2 in refluxing ethanol. Keywords: dihydropyridines, pyridine acylation. Electrophilic aromatic substitution reactions of pyridines are extraordinarily challenging. Instead of C-substitution at the pyridine ring, the electrophile typically forms an adduct with the pyridine nitrogen, which even further deactivates the already electron deficient pyridine ring toward electrophilic substitution. For example, the direct nitration of pyridine may require a reaction temperature of 330°C to provide only a 15% yield of 3-nitropyridine [1]. To the best of our knowledge, no direct, intermolecular C-acylations of pyridines have been reported [2]. This seriously limits the choice of methods for the preparation of the ubiquitous 3-substituted pyridines [3, 4]. In a limited number of cases, the lack of reactivity of pyridines toward electrophiles has been addressed by converting the recalcitrant pyridine into a temporarily activated 1,4-dihydropyridine [5-9]. In contrast to the electron poor parent pyridine, the electron rich 1,4-dihydropyridine features strongly enhanced reactivity toward electrophiles at the 3-position. Several steps are typically required including formation of the dihydropyridine, the subsequent reaction with an electrophile, and rearomatization to the desired 3-substituted pyridine. A similar concept has been ingeniously employed in a one pot nitration of pyridines in the presence of sulfite as the nucleophile that temporarily activates the pyridine, and then acts as a leaving group in an aromatization step [10, 11]. -

Chapter 18 Amines in Order for a Drug to Be Effective Orally, It Generally
Chapter 18 Amines In order for a drug to be effective orally, it generally has to be reasonable soluble in water so that it can be transported through the blood. Since amines are weak bases, they are often converted to salts with some acid and therefore may oral drugs have amine salts as part of their structure. One reason for their presence is that they confer some water solubility to the drug. The three-dimensional models show the shapes of amine molecules; notice the lone pair of electrons on nitrogen is not shown but affects the geometry about the nitrogen Primary, secondary and tertiary amines have 1, 2 or 3 alkyl groups attached to nitrogen. In these cases the alkyl group is the methyl In the IUPAC system, STEP 1 Name the longest carbon chain bonded to the N atoms as alkanamines by replacing e of the alkane name with amine. STEP 2 Number the carbon chain to locate the amine group and any substituents. N,N-Dimethylethanamine aminoethane 2-aminopropane 2-(N,N-dimethylamino)ethane 1-(N-methylamino)propane 2-(N-methylamino)butane Amines can also be names as groups attached to a hydrocarbon Aminobenzene is called aniline NH2 C H H C C C C H H C H NH2 NH2 NH CH3 Cl aniline 3-chloroaniline N-methylaniline aminobenzene 3-chloroaminobenzene N-methylaminobenzene Properties of amines The boiling points of amines are higher than alkanes of similar mass lower than alcohols of similar mass Amines are soluble in water if they have 1 to 5 carbon atoms; the N atom forms hydrogen bonds with the polar O—H bond in water An amine salt forms when an amine -
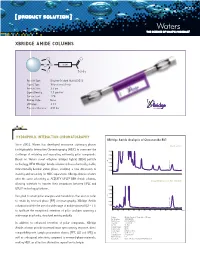
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

215-216 HH W12-Notes-Ch 15
Chem 215 F12 Notes Notes – Dr. Masato Koreeda - Page 1 of 17. Date: October 5, 2012 Chapter 15: Carboxylic Acids and Their Derivatives and 21.3 B, C/21.5 A “Acyl-Transfer Reactions” I. Introduction Examples: note: R could be "H" R Z R O H R O R' ester O carboxylic acid O O an acyl group bonded to R X R S acid halide* R' an electronegative atom (Z) thioester O X = halogen O R' R, R', R": alkyl, alkenyl, alkynyl, R O R' R N or aryl group R" amide O O O acid anhydride one of or both of R' and R" * acid halides could be "H" R F R Cl R Br R I O O O O acid fluoride acid chloride acid bromide acid iodide R Z sp2 hybridized; trigonal planar making it relatively "uncrowded" O The electronegative O atom polarizes the C=O group, making the C=O carbon "electrophilic." Resonance contribution by Z δ * R Z R Z R Z R Z C C C C O O O δ O hybrid structure The basicity and size of Z determine how much this resonance structure contributes to the hybrid. * The more basic Z is, the more it donates its electron pair, and the more resonance structure * contributes to the hybrid. similar basicity O R' Cl OH OR' NR'R" Trends in basicity: O weakest increasing basiciy strongest base base Check the pKa values of the conjugate acids of these bases. Chem 215 F12 Notes Notes –Dr. Masato Koreeda - Page 2 of 17. -

1. A. the First Reaction Is a Friedel-Crafts Acylation (FCA), Where the Major Product Is the Para- Isomer (60% Isolated Yield)
1. a. The first reaction is a Friedel-Crafts Acylation (FCA), where the major product is the para- isomer (60% isolated yield). The second reaction is a nitration, where the incoming electrophile (nitronium ion) is directed to the ortho position of the methoxy group. The last reaction is a Wolff-Kishner reduction that converts the acetyl group into an ethyl group. The nitro group does not react under these conditions. OCH OCH OCH OCH3 3 3 3 NO2 NO2 N2H4/KOH CH3COCl/AlCl3 H2SO4/HNO3 0 oC O CH 3 O CH3 CH3 (A) Reaction 1 (B) Reaction 2 (C) Reaction 3 (P) b. The best solvent for the FC-acylation is dichloromethane. Tetrahydrofuran is a fairly strong Lewis base, which would react and deactivate the AlCl3 catalyst. Ethanol would also react with AlCl3 and form alcoholates, which are inactive at FCA catalyst. Dichloromethane is polar enough to dissolve all three compounds but does not form adducts with AlCl3. Thus, aluminum chloride maintains its Lewis acidity. 3+ c. As discussed in lecture, AlCl3*6 H2O is not suitable as catalyst because the Al is not a strong Lewis acid anymore. In addition, larger amounts of water would destroy the acetyl chloride as well (=hydrolysis, CH3COCl + H2O ---- > CH3COOH + HCl). Consequently, the reaction would not proceed in the desired fashion. 3+ OH2 H2O OH2 Al H2O OH2 OH2 d. In order to determine the yield, one has to calculate the number of moles of the reactant and the product. nA = 1.90 mL * 0.996 g/mL/108.14 g/mol = 17.5 mmol nCH3COCl = 2.49 mL * 1.104 g/mL/78.5 g/mol = 35.0 mmol nAlCl3 = 4.67 g/133.5 g/mol = 35.0 mmol Compound (A) is the limiting reagent.