Research Collection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NVM Express and the PCI Express* SSD Revolution SSDS003
NVM Express and the PCI Express* SSD Revolution Danny Cobb, CTO Flash Memory Business Unit, EMC Amber Huffman, Sr. Principal Engineer, Intel SSDS003 Agenda • NVM Express (NVMe) Overview • New NVMe Features in Enterprise & Client • Driver Ecosystem for NVMe • NVMe Interoperability and Plugfest Plans • EMC’s Perspective: NVMe Use Cases and Proof Points The PDF for this Session presentation is available from our Technical Session Catalog at the end of the day at: intel.com/go/idfsessions URL is on top of Session Agenda Pages in Pocket Guide 2 Agenda • NVM Express (NVMe) Overview • New NVMe Features in Enterprise & Client • Driver Ecosystem for NVMe • NVMe Interoperability and Plugfest Plans • EMC’s Perspective: NVMe Use Cases and Proof Points 3 NVM Express (NVMe) Overview • NVM Express is a scalable host controller interface designed for Enterprise and client systems that use PCI Express* SSDs • NVMe was developed by industry consortium of 80+ members and is directed by a 13-company Promoter Group • NVMe 1.0 was published March 1, 2011 • Product introductions later this year, first in Enterprise 4 Technical Basics • The focus of the effort is efficiency, scalability and performance – All parameters for 4KB command in single 64B DMA fetch – Supports deep queues (64K commands per Q, up to 64K queues) – Supports MSI-X and interrupt steering – Streamlined command set optimized for NVM (6 I/O commands) – Enterprise: Support for end-to-end data protection (i.e., DIF/DIX) – NVM technology agnostic 5 NVMe = NVM Express NVMe Command Execution 7 1 -

Renewing Our Energy Future
Renewing Our Energy Future September 1995 OTA-ETI-614 GPO stock #052-003-01427-1 Recommended Citation: U.S. Congress, Office of Technology Assessment, Renewing Our Energy Fulture,OTA-ETI-614 (Washington, DC: U.S. Government Printing Office, September 1995). For sale by the U.S. Government Printing Office Superintendent of Documents, Mail Stop: SSOP, Washington, DC 20402-9328 ISBN 0-16 -048237-2 Foreword arious forms of renewable energy could become important con- tributors to the U.S. energy system early in the next century. If that happens, the United States will enjoy major economic, envi- ronmental, and national security benefits. However, expediting progress will require expanding research, development, and commer- cialization programs. If budget constraints mandate cuts in programs for renewable energy, some progress can still be made if efforts are focused on the most productive areas. This study evaluates the potential for cost-effective renewable energy in the coming decades and the actions that have to be taken to achieve the potential. Some applications, especially wind and bioenergy, are already competitive with conventional technologies. Others, such as photovol- taics, have great promise, but will require significant research and devel- opment to achieve cost-competitiveness. Implementing renewable energy will be also require attention to a variety of factors that inhibit potential users. This study was requested by the House Committee on Science and its Subcommittee on Energy and Environment; Senator Charles E. Grass- ley; two Subcommittees of the House Committee on Agriculture—De- partment Operations, Nutrition and Foreign Agriculture and Resource Conservation, Research and Forestry; and the House Subcommittee on Energy and Environment of the Committee on Appropriations. -

Intel® Optane™ Storage Performance and Implications on Testing Methodology October 27, 2017 Version 1.0
Intel® Optane™ Storage Performance and Implications on Testing Methodology October 27, 2017 Version 1.0 Executive Summary Since the unveiling in 2015, 3D XPoint™ Memory has shown itself to be the disruptive storage technology of the decade. Branded as Intel® Optane™ when packaged together with Intel’s storage controller and software, this new transistor-less solid-state ‘storage class memory’ technology promises lower latencies and increased system responsiveness previously unattainable from a non-volatile memory product. When coupled with NVMe and ever faster interfaces, Intel® Optane™ seeks to bridge the gap between slower storage and faster system RAM. Intel® Optane™ and 3D XPoint™ Technology 3D XPoint™ represents a radical departure from conventional non-volatile memory technologies. NAND flash memory stores bits by trapping an electrical charge within an insulated cell. Efficient use of die space mandates that programming be done by page and erasures by block. These limitations lead to a phenomenon called write amplification, where an SSD must manipulate relatively large chunks of data to achieve a given small random write operation, negatively impacting both performance and endurance. 3D XPoint™ is free of the block erase, page, and write amplification limitations inherent with NAND flash and can be in-place overwritten at the bit/byte/word level with no need for over- provisioning to maintain high random performance and consistency. 3D XPoint™ data access is more akin to that of RAM, and thanks to the significant reduction in write-related overhead compared to NAND, read responsiveness can be maintained even in the face of increased system write pressure. A deeper dive of how 3D XPoint™ Memory works is beyond the scope of this paper but can be found elsewhere on the web. -

Intel SSD 750 Series Evaluation Guide
Intel® Solid State Drive 750 Series Evaluation Guide September 2016 Order Number: 332075-002US Intel® Solid State Drive 750 Series Ordering Information Contact your local Intel sales representative for ordering information. Tests document performance of components on a particular test, in specific systems. Differences in hardware, software, or configuration will affect actual performance. Consult other sources of information to evaluate performance as you consider your purchase. Results have been estimated based on internal Intel analysis and are provided for informational purposes only. Any difference in system hardware or software design or configuration may affect actual performance. All documented performance test results are obtained in compliance with JESD218 Standards; refer to individual sub-sections within this document for specific methodologies. See www.jedec.org for detailed definitions of JESD218 Standards. Intel does not control or audit the design or implementation of third party benchmark data or Web sites referenced in this document. Intel encourages all of its customers to visit the referenced Web sites or others where similar performance benchmark data are reported and confirm whether the referenced benchmark data are accurate and reflect performance of systems available for purchase. The products described in this document may contain design defects or errors known as errata which may cause the product to deviate from published specifications. Current characterized errata are available on request. Contact your local Intel sales office or your distributor to obtain the latest specifications and before placing your product order. Intel and the Intel logo are trademarks of Intel Corporation in the U.S. and other countries. *Other names and brands may be claimed as the property of others. -

X-Series Hyperconverged Nodes Powered by Acuity 2.3 Setup & User Guide
X-Series Hyperconverged Nodes Powered by Acuity 2.3 Setup & User Guide Document Version 1.0 January 15, 2018 X-Series Hyperconverged Nodes Powered by Acuity Setup & User Guide, v1.0 Purpose This edition applies to Pivot3’s X-Series Hyperconverged Nodes powered by the Acuity software platform (Version 2.3.1 and above) and to any subsequent releases until otherwise indicated in new editions. Specific documentation on other Pivot3 offerings are available in the Pivot3 documentation repository. This document contains information proprietary to Pivot3, Inc. and shall not be reproduced or transferred to other documents or used for any purpose other than that for which it was obtained without the express written consent of Pivot3, Inc. How to Contact Pivot3 Pivot3, Inc. General Information: [email protected] 221 West 6th St., Suite 750 Sales: [email protected] Austin, TX 78701 Tech Support: [email protected] Tel: +1 512-807-2666 Website: www.pivot3.com Fax: +1 512-807-2669 Online Support: support.pivot3.com About This Guide This document provides procedures for: • Connecting X-Series Hyperconverged Nodes powered by Acuity to the VMware and ESXi management networks • Combining nodes to create an Acuity vPG This guide is written for Service Engineers and System Administrators configuring a Pivot3 X-Series Solution. The Pivot3 nodes should be set up as described in the appropriate hardware setup guide, and the procedures in this document require an understanding of the following: • The operating systems installed on the application servers connected to the Acuity vPG • Communication protocols installed on local application servers • vSphere Client for access to VMware ESXi hosts or vCenter © Copyright 2018 Pivot3, Inc. -

Selected Project Reports, Spring 2005 Advanced OS & Distributed Systems
Selected Project Reports, Spring 2005 Advanced OS & Distributed Systems (15-712) edited by Garth A. Gibson and Hyang-Ah Kim Jangwoo Kim††, Eriko Nurvitadhi††, Eric Chung††; Alex Nizhner†, Andrew Biggadike†, Jad Chamcham†; Srinath Sridhar∗, Jeffrey Stylos∗, Noam Zeilberger∗; Gregg Economou∗, Raja R. Sambasivan∗, Terrence Wong∗; Elaine Shi∗, Yong Lu∗, Matt Reid††; Amber Palekar†, Rahul Iyer† May 2005 CMU-CS-05-138 School of Computer Science Carnegie Mellon University Pittsburgh, PA 15213 †Information Networking Institute ∗Department of Computer Science ††Department of Electrical and Computer Engineering Abstract This technical report contains six final project reports contributed by participants in CMU’s Spring 2005 Advanced Operating Systems and Distributed Systems course (15-712) offered by professor Garth Gibson. This course examines the design and analysis of various aspects of operating systems and distributed systems through a series of background lectures, paper readings, and group projects. Projects were done in groups of two or three, required some kind of implementation and evalution pertaining to the classrom material, but with the topic of these projects left up to each group. Final reports were held to the standard of a systems conference paper submission; a standard well met by the majority of completed projects. Some of the projects will be extended for future submissions to major system conferences. The reports that follow cover a broad range of topics. These reports present a characterization of synchronization behavior -

HP MP9 G2 Retail System
QuickSpecs HP MP9 G2 Retail System Overview HP MP9 G2 Retail System FRONT/PORTS 1. Headphone Connector 4. USB 3.0 (charging) 2. Microphone or Headphone Connector 5. USB 3.0 (software selectable, default mode is microphone) 3. USB 3.0 Type-CTM 6. HDD indicator 7. Dual-State Power Button c04785658 — DA – 15369 Worldwide — Version 9 — January 2, 2017 Page 1 QuickSpecs HP MP9 G2 Retail System Overview REAR/PORTS 1. External antenna connector (antenna optional) 8. VGA monitor connector 2. Thumbscrew 9. DisplayPort (default, shown) or optional HDMI or serial 3. Padlock loop 10. (2) USB 3.0 ports (blue) 4. HP Keyed Cable Lock 11. (2) USB 3.0 ports (blue) allows for wake from S4/S5 with 5. External antenna connector (antenna optional) keyboard/mouse when connected and enabled in BIOS 6. Antenna cover 12. RJ-45 network connector 7. DisplayPort monitor connector 13. Power connector AT A GLANCE c04785658 — DA – 15369 Worldwide — Version 9 — January 2, 2017 Page 2 QuickSpecs HP MP9 G2 Retail System Overview Windows 10 IoT Enterprise for Retail (64-bit), Windows 10 Pro (64-bit), Windows Embedded 8.1 Industry Pro Retail (64- bit), Windows 8.1 Pro (64-bit), Windows Embedded Standard 7 (64-bit), Windows 7 Pro (32 & 64-bit), Windows Embedded POS Ready 7 (32 & 64-bit), FreeDOS UEFI BIOS developed and engineered by HP for better security, manageability and software image stability Intel® Q170 chipset Intel® 6th generation Core™ processors Intel® vPro™ Technology available with select processors Integrated Intel® HD Graphics Integrated Intel® i219LM -

Session Presentation
#CLUS Cisco HyperFlex Architecture Deep Dive, System Design and Performance Analysis Brian Everitt, Technical Marketing Engineer @CiscoTMEguy BRKINI-2016 #CLUS Agenda • Introduction • What’s New • System Design and Data Paths • Benchmarking Tools • Performance Testing • Best Practices • Conclusion #CLUS BRKINI-2016 © 2019 Cisco and/or its affiliates. All rights reserved. Cisco Public 3 About Me Brian Everitt • Technical Marketing Engineer with the CSPG BU • Focus on HyperFlex performance, benchmarking, quality and SAP solutions • 5 years as a Cisco Advanced Services Solutions Architect • Focus on SAP Hana appliances and TDI, UCS and storage #CLUS BRKINI-2016 © 2019 Cisco and/or its affiliates. All rights reserved. Cisco Public 4 Cisco Webex Teams Questions? Use Cisco Webex Teams to chat with the speaker after the session How 1 Find this session in the Cisco Live Mobile App 2 Click “Join the Discussion” 3 Install Webex Teams or go directly to the team space 4 Enter messages/questions in the team space Webex Teams will be moderated cs.co/ciscolivebot#BRKINI-2016 by the speaker until June 16, 2019. #CLUS © 2019 Cisco and/or its affiliates. All rights reserved. Cisco Public 5 Visit DC lounge outside WOS Grab a shirt, get a digital portrait for your social profiles, share your stories! Let’s Celebrate Visit the Data Center area inside 10 Years of WOS to see what’s new Unified Computing! Follow us! We made it simple. #CiscoDC https://bit.ly/2CXR33q You made it happen. #CLUS BRKINI-2016 © 2019 Cisco and/or its affiliates. All rights reserved. -

HP Proone 400 G1 All-In-One Business PC (21.5” Touch)
QuickSpecs HP ProOne 400 G1 All-in-One Business PC (21.5” Touch) Overview HP ProOne 400 G1 All-in-One Business PC FRONT 1. Microphones (optional) 2. Webcam activity LED 3. Webcam (optional) 4. Power button 5. Speakers DA - 14876 North America — Version 4 — March 20, 2014 Page 1 QuickSpecs HP ProOne 400 G1 All-in-One Business PC (21.5” Touch) Overview HP ProOne 400 G1 All-in-One Business PC BACK 1. Stand 2. Security screw 3. Power connector LED indicator 4. Power connector 5. DisplayPort 6. RJ-45 Gigabit Ethernet port 7. (4) USB 2.0 ports 8. Security lock slot 9. Stereo audio line out 10. Serial RS-232 port 11. VESA mount DA - 14876 North America — Version 4 — March 20, 2014 Page 2 QuickSpecs HP ProOne 400 G1 All-in-One Business PC (21.5” Touch) Overview DA - 14876 North America — Version 4 — March 20, 2014 Page 3 QuickSpecs HP ProOne 400 G1 All-in-One Business PC (21.5” Touch) Overview HP ProOne 400 G1 All-in-One Business PC SIDE 1. Optical Disc Drive (optional) 2. Optical eject button 3. Optical activity LED 4. Hard Disc Drive activity LED 5. Media Card Reader activity LED 6. SD Media Card Reader (optional) 7. (2) USB 3.0 Ports, including 1 fast charging port 8. Microphone jack 9. Headphone jack DA - 14876 North America — Version 4 — March 20, 2014 Page 4 QuickSpecs HP ProOne 400 G1 All-in-One Business PC (21.5” Touch) Overview At A Glance Windows 7 or Windows 8.1 21.5 inch Touch diagonal widescreen WLED backlit LCD Integrated all-in-one form factor Intel® H81 Express chipset Intel 4th Generation Core™ processors Integrated Intel HD Graphics -

Benchware Performance Suite Release 8.6 Release 8.6 Release 8.6
Exadata Evolution - Performance Baseline of X2, X3 and X4 Swiss Technical Exadata Community Meeting 19th of March 2014 Company Confidential Contents 1 Introduction 2 CPU Performance 3 Server Performance 4 Storage Performance 5 Database Load Performance 6 Summary copyright © 2014 by benchware.ch company confidential slide 2 Introduction Unpredictable performance of Oracle platforms – due to complexity Oracle application Oracle Benchware mission Application(s) . Quality assurance: calibrate Middleware efficiency of Oracle platforms . Platform evaluation: quantify price-performance ratio of Oracle Database System Fusion I/O Interconnect I/O Fusion platforms DataGuard File System platform Oracle Volume Manager . Capacity planning: deliver key Network O/S Network Storage Server performance metrics of Oracle platforms for capacity planning Storage System copyright © 2014 by benchware.ch company confidential slide 3 Introduction Holistic approach based on Key Performance Metrics Benchware methodology is based on readily understandable key performance metrics for . CPU performance . Server performance . Storage performance . Database performance for load, scan, OLTP transactions, . Benchware uses a set of representative Oracle benchmark tests to calibrate key performance metrics of all platform components This presentation shows some of these benchmark results copyright © 2014 by benchware.ch company confidential slide 4 Introduction Key Performance Metrics should be self-explanatory Source: www.bmw.de copyright © 2014 by benchware.ch company -

Accelerating Instant Recovery
Exploring The True Cost of Converged vs. Hyperconverged Infrastructure Accelerating Instant Recovery: Cohesity Outperforms Veeam-Based System A DeepStorage Technology Validation Report i Prepared for Cohesity Accelerating Instant Recovery: Cohesity Outperforms Veeam-Based System About DeepStorage DeepStorage, LLC. is dedicated to revealing the deeper truth about storage, networking and related data center technologies to help information technology professionals deliver superior services to their users and still get home at a reasonable hour. DeepStorage Reports are based on our hands-on testing and over 30 years of experience making technology work in the real world. Our philosophy of real world testing means we configure systems as we expect most cus- tomers will use them thereby avoiding “Lab Queen” configurations designed to maximize benchmark performance. This report was sponsored by our client. However, DeepStorage always retains final edi- torial control over our publications. Copyright © 2018 DeepStorage, LLC. All rights reserved worldwide. ii Accelerating Instant Recovery: Cohesity Outperforms Veeam-Based System Contents About DeepStorage ii Introduction 1 The Bottom Line 1 Direct Recovery 2 The Cohesity DataPlatform 2 Cohesity System Configuration 4 Hardware Specs 4 The Scenario – Build Vs Buy 4 Our Internally Engineered Veeam System 4 Comparing The Scale-Out Architectures 5 The Tests 5 The Workloads 6 HammerDB TPC-C “like” workload 6 The Backup Phase 7 The Results 7 Basic Backup/ Restore Performance 7 Direct Recovery Performance -
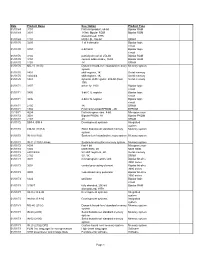
Date Product Name Description Product Type 01/01/69 3101 First
Date Product Name Description Product Type 01/01/69 3101 first Intel product, 64-bit Bipolar RAM 01/01/69 3301 1K-bit, Bipolar ROM Bipolar ROM discontinued, 1976 01/01/69 1101 MOS LSI, 256-bit SRAM 01/01/70 3205 1-of-8 decoder Bipolar logic circuit 01/01/70 3404 6-bit latch Bipolar logic circuit 01/01/70 3102 partially decoded, 256-bit Bipolar RAM 01/01/70 3104 content addressable, 16-bit Bipolar RAM 01/01/70 1103 1K DRAM 01/01/70 MU-10 (1103) Dynamic board-level standard memory Memory system system 01/01/70 1401 shift register, 1K Serial memory 01/01/70 1402/3/4 shift register, 1K Serial memory 01/01/70 1407 dynamic shift register, 200-bit (Dual Serial memory 100) 01/01/71 3207 driver for 1103 Bipolar logic circuit 01/01/71 3405 3-bit CTL register Bipolar logic circuit 01/01/71 3496 4-bit CTL register Bipolar logic circuit 01/01/71 2105 1K DRAM 01/01/71 1702 First commercial EPROM - 2K EPROM 11/15/71 4004 first microprocessor, 4-bit Microprocessor 01/01/72 3601 Bipolar PROM, 1K Bipolar PROM 01/01/72 2107 4K DRAM 01/01/72 SIM 4, SIM 8 Development systems Integrated system 01/01/72 CM-50 (1101A) Static board-level standard memory Memory system system 01/01/72 IN-10 (1103) System-level standard memory system Memory system 01/01/72 IN-11 (1103) Univac System-level custom memory system Memory system 01/01/72 8008 first 8-bit Microprocessor 01/01/72 1302 MOS ROM, 2K MOS ROM 01/01/72 2401/2/3/4 5V shift registers, 2K Serial memory 01/01/72 2102 5V, 1K SRAM 01/01/73 3001 microprogram control unit Bipolar bit-slice 3000 series 01/01/73 3002 central