Clode-BSC-2016-Ocula
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intraocular Pressure Rise After Phacoemulsification Prophylactic
British Journal of Ophthalmology 1995; 79: 809-813 809 Intraocular pressure rise after phacoemulsification Br J Ophthalmol: first published as 10.1136/bjo.79.9.809 on 1 September 1995. Downloaded from with posterior chamber lens implantation: effect of prophylactic medication, wound closure, and surgeon's experience Thomas G Bomer, Wolf-Dietrich A Lagreze, Jens Funk Abstract pressure rises usually occur between 6 and 8 Aims-A prospective clinical trial was hours after surgery.6 carried out to evaluate the effect of Various antiglaucomatous agents have been prophylactic medication, the technique of used to prevent the intraocular pressure rise wound closure, and the surgeon's experi- after cataract extraction. Oral acetazolamide15 16 ence on the intraocular pressure rise after and topical timolol'6-19 lowered the pressure cataract extraction. rise in the early period after intracapsular and Methods-In 100 eyes, the intraocular extracapsular cataract extraction. Levobunolol pressure was measured before as well as proved to be superior to timolol 4-7 hours 2-4, 5-7, and 22-24 hours after phaco- after extracapsular cataract extraction.20 Apra- emulsification and posterior chamber lens clonidine lowered the intraocular pressure rise implantation. Each of 25 patients received after uncomplicated phacoemulsification21 and either 1% topical apraclonidine, 0.5%/o extracapsular cataract extraction22 23 when topical levobunolol, 500 mg oral acetazo- given 30 minutes to 1 hour before surgery, lamide, or placebo. Forty four eyes were whereas immediate postoperative treatment operated with sclerocorneal sutureless with apraclonidine was ineffective.22 24 Miotics tunnel and 56 eyes with corneoscleral are frequently used to promote miosis and incision and suture. -

The Effect of Topical Beta-Adrenoceptor Blocking Agents on Pulsatile Ocular Blood Flow
THE EFFECT OF TOPICAL BETA-ADRENOCEPTOR BLOCKING AGENTS ON PULSATILE OCULAR BLOOD FLOW C. D. MORSMAN, M. E. BOSEM, M. LUSKY and R. N. WEINREB San Diego, California SUMMARY factors (e.g. hypertension, diabetes, peripheral Thirty-three ocular hypertensive patients (21 with vascular disease and vasospasm) to the vascular primary open angle glaucoma and 12 glaucoma system suggest that blood flow in the optic nerve suspects) were randomly assigned to receive either head and retina may be altered in glaucoma.4 Of the timolol, levobunolol or betaxolol in one eye. Pulsatile various vascular beds within the posterior segment, ocular blood flow (POBF) was measured before the choroidal circulation is of particular interest since treatment (baseline) and 2 hours after drop adminis it provides the major contribution to the blood flow tration. After 1 week of regular twice-daily dosage, of the optic nerve at the level of the lamina cribrosa.5 POBF was measured again both immediately before Beta-2 adrenergic receptors have been demon and 2 hours after drop instillation. All measurements strated in human optic nerve,6 as well as in choroidal were made by an investigator masked to treatment. and retinal blood vessels.7 Blockade of these POBF increased by 11% (p = 0.09) at week 0 after receptors can cause vasoconstrictionS which could levobunolol administration, and by 22% (p = 0.20) at adversely affect visual function if adequate concen week 1 before drop administration compared with trations of the drug diffused into the posterior baseline. It dropped by 23% and 25% (p = 0.04 and segment of the eye or were absorbed systemically. -

Brimonidine Tartrate; Brinzolamide
Contains Nonbinding Recommendations Draft Guidance on Brimonidine Tartrate ; Brinzolamide This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA, or the Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the Office of Generic Drugs. Active Ingredient: Brimonidine tartrate; Brinzolamide Dosage Form; Route: Suspension/drops; ophthalmic Strength: 0.2%; 1% Recommended Studies: One study Type of study: Bioequivalence (BE) study with clinical endpoint Design: Randomized (1:1), double-masked, parallel, two-arm, in vivo Strength: 0.2%; 1% Subjects: Males and females with chronic open angle glaucoma or ocular hypertension in both eyes. Additional comments: Specific recommendations are provided below. ______________________________________________________________________________ Analytes to measure (in appropriate biological fluid): Not applicable Bioequivalence based on (95% CI): Clinical endpoint Additional comments regarding the BE study with clinical endpoint: 1. The Office of Generic Drugs (OGD) recommends conducting a BE study with a clinical endpoint in the treatment of open angle glaucoma and ocular hypertension comparing the test product to the reference listed drug (RLD), each applied as one drop in both eyes three times daily at approximately 8:00 a.m., 4:00 p.m., and 10:00 p.m. for 42 days (6 weeks). 2. Inclusion criteria (the sponsor may add additional criteria): a. Male or nonpregnant females aged at least 18 years with chronic open angle glaucoma or ocular hypertension in both eyes b. -

Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg
OPTOMETRIC STUDY CENTER Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg. % Product Sizes Side Effects Warnings Reduction CHOLINERGIC AGENTS Direct Pilocarpine (generic) Pilocarpine 1%, 2%, 4% Increases trabecular outflow BID-QID/15-25% 15ml Headache, blurred vision, myopia, retinal detachment, bronchiole constriction, Angle closure, shortness of breath, retinal narrowing of angle detachment Indirect Phospholine Iodide (Pfizer) Echothiophate iodide 0.125% Increases trabecular outflow QD-BID/15-25% 5ml Same as above plus cataractogenic iris cysts in children, pupillary block, Same as above, plus avoid prior to any increased paralysis with succinylcholine general anesthetic procedure ALPHA-2 AGONISTS Alphagan P (Allergan) Brimonidine tartrate 0.1%, 0.15% with Purite Decreases aqueous production, increases BID-TID/up to 26% 5ml, 10ml, 15ml Dry mouth, hypotension, bradycardia, follicular conjunctivitis, ocular irritation, Monitor for shortness of breath, dizziness, preservative uveoscleral outflow pruritus, dermatitis, conjunctival blanching, eyelid retraction, mydriasis, drug ocular redness and itching, fatigue allergy Brimonidine tartrate Brimonidine tartrate 0.15%, 0.2% Same as above Same as above 5ml, 10ml Same as above Same as above (generic) Iopidine (Novartis) Apraclonidine 0.5% Decreases aqueous production BID-TID/up to 25% 5ml, 10ml Same as above but higher drug allergy (40%) Same as above BETA-BLOCKERS Non-selective Betagan (Allergan) Levobunolol 0.25%, 0.5% Decreases -

Use of Emulsions for Intra- and Periocular Injection
(19) & (11) EP 1 611 879 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 9/107 (2006.01) 12.08.2009 Bulletin 2009/33 (21) Application number: 04291684.1 (22) Date of filing: 02.07.2004 (54) Use of emulsions for intra- and periocular injection Verwendung von Emulsionen zur intra- und periocularen Injection Utilisation des émulsions pour injection intra- et périoculaire. (84) Designated Contracting States: (74) Representative: de Mareüil-Villette, Caroline et al AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Cabinet Plasseraud HU IE IT LI LU MC NL PL PT RO SE SI SK TR 52 rue de la Victoire 75440 Paris Cedex 09 (FR) (43) Date of publication of application: 04.01.2006 Bulletin 2006/01 (56) References cited: EP-A- 0 521 799 EP-A- 1 020 194 (73) Proprietors: WO-A-02/09667 WO-A-93/18852 • Novagali Pharma S.A. WO-A-94/05298 WO-A-03/053405 91000 Evry (FR) US-A- 5 632 984 • CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE (CNRS) • KLANG S H ET AL: "PHYSICOCHEMICAL 75016 Paris (FR) CHARACTERIAZATION AND ACUTE TOXICITY • INSTITUT NATIONAL DE LA SANTE ET DE LA EVALUATION OF A POSITIVELY-CHARGED RECHERCHE MEDICALE (INSERM) SUBMICRON EMULSION VEHICLE" JOURNAL 75013 Paris (FR) OF PHARMACY AND PHARMACOLOGY, • YISSUM RESEARCH DEVELOPMENT COMPANY LONDON, GB, vol. 46, no. 12, December 1994 OF THE HEBREW UNIVERSITY OF JERUSALEM (1994-12), pages 986-993, XP008005426 ISSN: 91390 Jerusalem (IL) 0022-3573 • KLANG S ET AL: "INFLUENCE OF EMULSION (72) Inventors: DROPLET SURFACE CHARGE ON • Rabinovich-Guilatt, Laura INDOMETHACIN OCULAR TISSUE 75015 Paris (FR) DISTRIBUTION" PHARMACEUTICAL • De Kozak, Yvonne DEVELOPMENT AND TECHNOLOGY, NEW 75013 Paris (FR) YORK, NY, US, vol. -

AZARGA, INN-Brinzolamide/Timolol
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT AZARGA 10 mg/ml + 5 mg/ml eye drops, suspension 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One ml of suspension contains 10 mg brinzolamide and 5 mg timolol (as timolol maleate). Excipient with known effect: One ml of suspension contains 0.10 mg benzalkonium chloride. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Eye drops, suspension (eye drops) White to off-white uniform suspension, pH 7.2 (approximately). 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Decrease of intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension for whom monotherapy provides insufficient IOP reduction (see section 5.1). 4.2 Posology and method of administration Posology Use in adults, including the elderly The dose is one drop of AZARGA in the conjunctival sac of the affected eye(s) twice daily. When using nasolacrimal occlusion or closing the eyelids, the systemic absorption is reduced. This may result in a decrease in systemic side effects and an increase in local activity (see section 4.4). If a dose is missed, treatment should be continued with the next dose as planned. The dose should not exceed one drop in the affected eye (s) twice daily. When substituting another ophthalmic antiglaucoma medicinal product with AZARGA, the other medicinal product should be discontinued and AZARGA should be started the following day. Special populations Paediatric population The safety and efficacy of AZARGA in children and adolescents aged 0 to 18 years have not yet been established. -

Fluid Ophthalmic Composition Based on Lipid Microparticles Containing at Least One Active Principle
Europaisches Patentamt J European Patent Office Office europden des brevets (11) Publication number : 0 437 368 A1 EUROPEAN PATENT APPLICATION (21) Application number: 91300181.4 ® int. ci.5 : A61K 9/06, A61K 9/16 @ Date of filing : 10.01.91 © Priority : 12.01.90 FR 9000340 (72) Inventor : Rozier, Annouk 23 Bd Lafayette F-63000 Clermont-Ferrand (FR) @ Date of publication of application : 17.07.91 Bulletin 91/29 74) Representative : Hesketh, Alan, Dr. et al European Patent Department Merck & Co., @ Designated Contracting States : Inc. Tertings Park Eastwick Road CH DE FR GB IT LI NL Harlow Essex, CM20 2QR (GB) © Applicant : LABORATOIRES MERCK, SHARP & DOHME-CHIBRET 3, Avenue Hoche F-75008 Paris (FR) (S) Fluid ophthalmic composition based on lipid microparticles containing at least one active principle. (57) There is described a fluid ophthalmic composition which comprises a suspension in a fluid dispersant medium of lipid microparticles containing at least one active principle. The composition enables improved availability of the active principle to be obtained as a result of high intraocular levels. 00 <0 CO Q. UJ Jouve, 18, rue Saint-Denis, 75001 PARIS EP 0 437 368 A1 FLUID OPHTHALMIC COMPOSITION BASED ON LIPID MICROPARTICLES CONTAINING AT LEAST ONE ACTIVE PRINCIPLE The present invention relates to a fluid ophthalmic composition. Many ophthalmic compositions are currently available in liquid or solid form, but none of them is, in fact, completely satisfactory. In effect, liquid ophthalmic compositions, although easy to use, have some drawbacks ; in particular, it is 5 difficult to obtain a sustained or delayed action of the active principle which they contain. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -
Guidance for the Format and Content of the Protocol of Non-Interventional
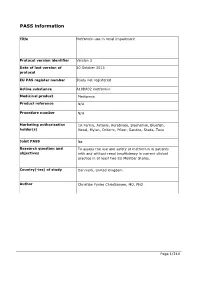
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (Apraclonidine Hydrochloride) Eye Drops 0.5%
NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (apraclonidine hydrochloride) Eye Drops 0.5%. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each mL of Iopidine Eye Drops 0.5% contains apraclonidine hydrochloride 5.75 mg, equivalent to apraclonidine base 5 mg. Excipient with known effect Benzalkonium chloride 0.1 mg per 1 mL as a preservative. For the full list of excipients, see section 6.1. 2. PHARMACEUTICAL FORM Eye drops, solution, sterile, isotonic. 4. CLINICAL PARTICULARS 4.1. Therapeutic indications Iopidine Eye Drops 0.5% are indicated for short-term adjunctive therapy in patients on maximally tolerated medical therapy who require additional IOP reduction. Patients on maximally tolerated medical therapy who are treated with Iopidine Eye Drops 0.5% to delay surgery should have frequent follow up examinations and treatment should be discontinued if the intraocular pressure rises significantly. The addition of Iopidine Eye Drops 0.5% to patients already using two aqueous suppressing drugs (i.e. beta-blocker plus carbonic anhydrase inhibitor) as part of their maximally tolerated medical therapy may not provide additional benefit. This is because apraclonidine is an aqueous-suppressing drug and the addition of a third aqueous suppressant may not significantly reduce IOP. The IOP-lowering efficacy of Iopidine Eye Drops 0.5% diminishes over time in some patients. This loss of effect, or tachyphylaxis, appears to be an individual occurrence with a variable time of onset and should be closely monitored. The benefit for most patients is less than one month. 4.2. Dose and method of administration Dose One drop of Iopidine Eye Drops 0.5% should be instilled into the affected eye(s) three times per day. -

Canine Red Eye Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO
PEER REVIEWED Clinical Approach to the CANINE RED EYE Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO he acute red eye is a common clinical challenge for tion of the deep episcleral vessels, and is characterized general practitioners. Redness is the hallmark of by straight and immobile episcleral vessels, which run Tocular inflammation; it is a nonspecific sign related 90° to the limbus. Episcleral injection is an external to a number of underlying diseases and degree of redness sign of intraocular disease, such as anterior uveitis and may not reflect the severity of the ocular problem. glaucoma (Figures 3 and 4). Occasionally, episcleral Proper evaluation of the red eye depends on effective injection may occur in diseases of the sclera, such as and efficient diagnosis of the underlying ocular disease in episcleritis or scleritis.1 order to save the eye’s vision and the eye itself.1,2 • Corneal Neovascularization » Superficial: Long, branching corneal vessels; may be SOURCE OF REDNESS seen with superficial ulcerative (Figure 5) or nonul- The conjunctiva has small, fine, tortuous and movable vessels cerative keratitis (Figure 6) that help distinguish conjunctival inflammation from deeper » Focal deep: Straight, nonbranching corneal vessels; inflammation (see Ocular Redness algorithm, page 16). indicates a deep corneal keratitis • Conjunctival hyperemia presents with redness and » 360° deep: Corneal vessels in a 360° pattern around congestion of the conjunctival blood vessels, making the limbus; should arouse concern that glaucoma or them appear more prominent, and is associated with uveitis (Figure 4) is present1,2 extraocular disease, such as conjunctivitis (Figure 1). -

WO 2014/066775 Al 1 May 2014 (01.05.2014) W P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/066775 Al 1 May 2014 (01.05.2014) W P O PCT (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61F 9/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 13/066834 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 25 October 2013 (25.10.201 3) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (25) Filing Language: English OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (26) Publication Language: English SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (30) Priority Data: ZW. 61/719,144 26 October 2012 (26. 10.2012) US (84) Designated States (unless otherwise indicated, for every (71) Applicant: FORSIGHT VISION5, INC. [US/US]; 191 kind of regional protection available): ARIPO (BW, GH, Jefferson Drive, Menlo Park, CA 94025 (US). GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, (72) Inventors: RUBIN, Anne, Brody; 191 Jefferson Drive, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Menlo Park, CA 94025 (US).