Presented to the Faculty of the Graduate Division by Robert John Rosscup in Partial Fulfillment of the Requirements for the Degr
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Immersion Cooling of Electronics in Dod Installations
LBNL-1005666 Immersion Cooling of Electronics in DoD Installations Henry Coles and Magnus Herrlin Energy Technologies Area May 2016 DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor The Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or The Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or The Regents of the University of California. ABSTRACT A considerable amount of energy is consumed to cool electronic equipment in data centers. A method for substantially reducing the energy needed for this cooling was demonstrated. The method involves immersing electronic equipment in a non-conductive liquid that changes phase from a liquid to a gas. The liquid used was 3M Novec 649. Two-phase immersion cooling using this liquid is not viable at this time. The primary obstacles are IT equipment failures and costs. However, the demonstrated technology met the performance objectives for energy efficiency and greenhouse gas reduction. -

Working with Hazardous Chemicals
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -
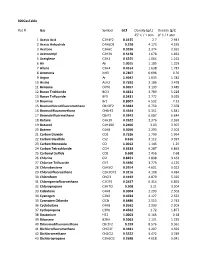
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Environmental Health and Safety Vacuum Traps
Environmental Health and Safety Vacuum Traps Always place an appropriate trap between experimental apparatus and the vacuum source. The vacuum trap: • protects the pump, pump oil and piping from the potentially damaging effects of the material; • protects people who must work on the vacuum lines or system, and; • prevents vapors and related odors from being emitted back into the laboratory or system exhaust. Improper trapping can allow vapor to be emitted from the exhaust of the vacuum system, resulting in either reentry into the laboratory and building or potential exposure to maintenance workers. Proper traps are important for both local pumps and building systems. Proper Trapping Techniques To prevent contamination, all lines leading from experimental apparatus to the vacuum source must be equipped with filtration or other trapping as appropriate. • Particulates: use filtration capable of efficiently trapping the particles in the size range being generated. • Biological Material: use a High Efficiency Particulate Air (HEPA) filter. Liquid disinfectant (e.g. bleach or other appropriate material) traps may also be required. • Aqueous or non-volatile liquids: a filter flask at room temperature is adequate to prevent liquids from getting to the vacuum source. • Solvents and other volatile liquids: use a cold trap of sufficient size and cold enough to condense vapors generated, followed by a filter flask capable of collecting fluid that could be aspirated out of the cold trap. • Highly reactive, corrosive or toxic gases: use a sorbent canister or scrubbing device capable of trapping the gas. Environmental Health and Safety 632-6410 January 2010 EHSD0365 (01/10) Page 1 of 2 www.stonybrook.edu/ehs Cold Traps For most volatile liquids, a cold trap using a slush of dry ice and either isopropanol or ethanol is sufficient (to -78 deg. -

A Sample AMS Latex File
Li, L. et al. (2021): JoSS, Vol. 10, No. 1, pp. 983–993 (Peer-reviewed article available at www.jossonline.com) www.adeepakpublishing.com www. JoSSonline.com Preliminary Thermal Validation Tests for Education-Class CubeSats and Weather- Balloon Payloads Lingqi Li and Kenjiro S. Lay Penn State University State College, PA, US Masataka Okutsu Penn State University Abington Abington, PA, US Abstract Low development and launch costs of CubeSats, a type of small spacecraft typically one to three liters in volume, have made space science accessible to educational institutions, offering engaging opportunities for stu- dents in the science, technology, engineering, and mathematics (STEM) disciplines. Some university teams work- ing on these education-class CubeSats conduct high-altitude flight experiments using balloons to test their instru- ments in the harsh environment at the edges of the troposphere and the stratosphere. Whether for the balloon experiment or for the actual spaceflight, temperatures of the operating environments are of concern. Instruments flown in space must be qualified for wide thermal ranges (e.g., −40°C to 70°C) in vacuum conditions. Likewise, instruments flown on the balloons must be able to operate in a similarly large range of temperatures (e.g., −50°C to 50°C) in the reduced pressure environment. Unfortunately, a thermal-vacuum chamber—standard testing equipment for spacecraft—is not accessible to many university teams. This paper presents incubator testing and cooling-bath testing methods as preliminary thermal validation tests that may be carried out easily, safely, and inexpensively, without any need for the expensive thermal-vacuum chamber. We also discuss an add-on demon- stration in which a CubeSat prototype was flown on a weather balloon to an altitude of ~16 km. -

Rapid Cryogenic Fixation of Biological Specimens for Electron Microscopy
RAPID CRYOGENIC FIXATION OF BIOLOGICAL SPECIMENS FOR ELECTRON MICROSCOPY KEITH PATRICKRYAN A thesis submitted in partial fulfilment of the requirements of the Council for National Academic Awards for the degree of Doctor of Philosophy September 1991 Polytechnic South West in collaboration with the Marine Biological Association of the United Kingdom and Plymouth Marine Laboratory ----- . \ ~ ,, '' - .... .._~ .. ·=·~-·-'-·-'" --······ --~....... ~=.sn.-.......... .r.=-..-> POL VTECHNIC SOUTH WEST liBRARY SERVICES Item C!O 00 7 9 4 9 3-0 No. ', )'; I Class 1 !) -, C!f -RYA !No. rr ... · ,Contl No. :x70ZS\0253 ·. ' COPYRIGHT This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without the authors prior written consent. 2 CONTENTS Page List of Figures 8 List of Tables 10 Abstract 11 Acknowledgements 12 1 Introduction 13 2 Literature Review 23 2.1 Background to specimen preservation for microscopy 23 2.2 Problems of chemical processing for electron rhlcroscopy 23 2.3 Introduction of cryotechniques into microscopy methods 24 2.4 The potential of cryofixation 25 2.5 The problems of cryofixation 25 2.6 Water, cooling and crystal nucleation 26 2. 7 Cell water 29 2.8 Crystallisation and latent heat release 29 2.9 Phase separation and eutectic temperature 30 2.10 Types of ice 31 2.11 Phase transitions 32 2.12 Ice crystal growth in frozen specimens after freezing 34 2.13 Cryoprotection against ice crystal damage 37 2.14 Modelling the cooling process 38 2.15 Cooling methods 45 2.16 Coolants (liquid) 46 2.17 Coolants (solid) 46 2.18 Plunge cooling methods 48 2.19 Jet cooling methods 51 2.20 Cryoblock methods 54 2.21 Rapid cooling experiments 59 3 2.22 Specimen rewarming during handling after freezing 74 2.23 Conclusions 74 3. -

(12) United States Patent (10) Patent No.: US 8,911,640 B2 Nappa Et Al
USOO891. 1640B2 (12) United States Patent (10) Patent No.: US 8,911,640 B2 Nappa et al. (45) Date of Patent: Dec. 16, 2014 (54) COMPOSITIONS COMPRISING 5,736,063 A 4/1998 Richard et al. FLUOROOLEFNS AND USES THEREOF 5,744,052 A 4/1998 Bivens 5,788,886 A 8, 1998 Minor et al. 5,897.299 A * 4/1999 Fukunaga ..................... 417.316 (71) Applicant: E I du Pont de Nemours and 5,969,198 A 10/1999 Thenappan et al. Company, Wilmington, DE (US) 6,053,008 A 4/2000 Arman et al. 6,065.305 A 5/2000 Arman et al. (72) Inventors: Mario Joseph Nappa, Newark, DE 6,076,372 A 6/2000 Acharya et al. 6,111,150 A 8/2000 Sakyu et al. (US); Barbara Haviland Minor, Elkton, 6,176,102 B1 1/2001 Novak et al. MD (US); Allen Capron Sievert, 6,258,292 B1 7/2001 Turner Elkton, MD (US) 6,300,378 B1 10/2001 Tapscott 6.426,019 B1 7/2002 Acharya et al. (73) Assignee: E I du Pont de Nemours and 6,610,250 B1 8, 2003 Tuma Company, Wilmington, DE (US) 6,858,571 B2 2/2005 Pham et al. 6,969,701 B2 11/2005 Singh et al. 7,708,903 B2 5, 2010 Sievert et al. *) Notice: Subject to anyy disclaimer, the term of this 8,012,368 B2 9/2011 Nappa et al. patent is extended or adjusted under 35 8,070,976 B2 12/2011 Nappa et al. U.S.C. 154(b) by 0 days. -

Solid-Liquid Phase Equilibria and Crystallization of Disubstituted Benzene Derivatives
Royal Institute of Technology School of Chemical Science and Engineering Department of Chemical Engineering and Technology Division of Transport Phenomena Solid-Liquid Phase Equilibria and Crystallization of Disubstituted Benzene Derivatives Fredrik Nordström Doctoral Thesis Akademisk avhandling som med tillstånd av Kungliga Tekniska Högskolan i Stockholm framlägges till offentlig granskning för avläggande av teknologie doktorsexamen den 30:e Maj 2008, kl. 10:00 i sal D3, Lindstedtsvägen 5, Stockholm. Avhandlingen försvaras på engelska. i Cover picture: Crystals of o-hydroxybenzoic acid (salicylic acid) obtained through evaporation crystallization in solutions of ethyl acetate at around room temperature. Solid-Liquid Phase Equilibria and Crystallization of Disubstituted Benzene Derivatives Doctoral Thesis © Fredrik L. Nordström, 2008 TRITA-CHE Report 2008-32 ISSN 1654-1081 ISBN 978-91-7178-949-5 KTH, Royal Institute of Technology School of Chemical Science and Engineering Department of Chemical Engineering and Technology Division of Transport Phenomena SE-100 44 Stockholm Sweden Paper I: Copyright © 2006 by Wiley InterScience Paper II: Copyright © 2006 by Elsevier Science Paper III: Copyright © 2006 by the American Chemical Society Paper IV: Copyright © 2006 by the American Chemical Society ii In loving memory of my grandparents Aina & Vilmar Nordström iii i v Abstract The Ph.D. project compiled in this thesis has focused on the role of the solvent in solid-liquid phase equilibria and in nucleation kinetics. Six organic substances have been selected as model compounds, viz. ortho-, meta- and para-hydroxybenzoic acid, salicylamide, meta- and para-aminobenzoic acid. The different types of crystal phases of these compounds have been explored, and their respective solid-state properties have been determined experimentally. -

Heating and Cooling Chemical Mixtures Revision Date: 11/01/19 Prepared By: Michael Roy P.I.: Prof
Section 5.7 Title: Heating and Cooling Chemical Mixtures Revision Date: 11/01/19 Prepared By: Michael Roy P.I.: Prof. John F. Berry Prior Approval: This procedure is NOT considered hazardous enough that prior approval is needed from the Principal Investigator. Involves Use of Particularly Hazardous Substance (PHS)? No ___ Carcinogen ___ Reproductive Toxin ___ High Acute Toxicity Does this procedure require medical surveillance? No Does this require use of a fit-tested respirator? No Brief Description of Procedure: Overview of common heating and cooling methods used in the Berry labs. Location: List the locations (buildings/rooms) where this procedure may be performed. For use of a PHS indicate a more precise location within the room, if appropriate, as a designated area. Daniels Chemistry - All Berry group labs Chemicals Involved: Chemical Physical or Health Hazard (e.g. carcinogen, corrosive) Organic solvents (cooling) Consult relevant SDSs for more details Dry ice Frostbite Liquid nitrogen Frostbite, asphyxiation Other Hazards: Include hazards, other than chemical, that may be present during operation of the procedure. Burns (heating) and frostbite (cooling). Exposure Controls: (Check all that apply) PPE: _X_ Safety Glasses ___ Face Shield ___Chemical Splash Goggles ___Chemical Apron _X_ Gloves (Nitrile) _X_ Lab Coat ___Respirator (type) ___Other: Engineering Controls: _X_ Fume Hood ___Biosafety Cabinet ___ Glove box ___ Vented gas cabinet ___Other: Administrative Controls: List any specific work practices needed to perform this procedure (e.g., cannot be performed alone, must notify other staff members before beginning, etc.). N/A Task Hazard Control Table: For procedures involving numerous steps, it may be convenient to indicate specific requirements for individual tasks in the table below: N/A Waste Disposal: Describe any chemical waste generated and the disposal method used. -

Chemical Laboratory Techniques - Gelosa D
FUNDAMENTALS OF CHEMISTRY – Vol. I - Chemical Laboratory Techniques - Gelosa D. and Sliepcevich A. CHEMICAL LABORATORY TECHNIQUES Gelosa D. and Sliepcevich A. Politecnico di Milano, Italy Keywords: batch reactor, boiling point, boiling stones, bubble-plate columns, continuous reactor, cooling bath, crystallization, drying, electric hot plate, electric mantle, equipment, extraction, filtration, fractional distillation, glass joint, glassware, heating bath, laboratory, nonpolar solvents, packed columns, polar solvents, purification, separation, simple distillation, steam distillation, stirring, technique, tubular reactor, vacuum distillation, vacuum pump. Contents 1. Common Laboratory Apparatus 1.1. Glassware 1.2. Heating of the Reaction Mixture 1.2.1. Burners 1.2.2. Heating Baths 1.2.3. Electric Hot Plates and Electric Heating Mantles 1.3. Cooling of the Reaction Mixtures 1.4. Stirring 2. The Reaction 2.1. Batch Reactors 2.2. Continuous Stirred-Tank Reactors 2.3. Tubular Reactors 2.3.1. Pulse Reactors 3. Isolation and Purification Techniques 3.1. Filtration 3.2. Extraction 3.2.1. Liquid–Liquid Extraction 3.2.2. Liquid–Solid Extraction 3.3. Crystallization 3.4. Distillation 3.4.1. TheoreticalUNESCO Aspects – EOLSS 3.4.2. Simple Distillation 3.4.3. Fractional Distillation 3.4.4. Distillation under Reduced Pressure 3.4.5. Steam DistillationSAMPLE CHAPTERS Glossary Bibliography Biographical Sketches Summary Basic laboratory techniques in chemistry have an essential place in the training of a chemist. They provide a good background for experimental skills and for subsequent ©Encyclopedia of Life Support Systems (EOLSS) FUNDAMENTALS OF CHEMISTRY – Vol. I - Chemical Laboratory Techniques - Gelosa D. and Sliepcevich A. independent research. In this chapter, first of all, we shall consider some of the standard pieces of glassware and equipment that you will use in the laboratory. -

Environmental Health Criteria 166 METHYL BROMIDE
Environmental Health Criteria 166 METHYL BROMIDE Please note that the layout and pagination of this web version are not identical with the printed version. Methyl Bromide (EHC 166, 1995) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 166 METHYL BROMIDE This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. First draft prepared by Dr. R.F. Hertel and Dr. T. Kielhorn. Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1995 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, -

Status of Numerical Modeling of Halocarbon Impacts on Stratospheric Ozone
THE STATUS OF NUMERICAL MODELING OF HALOCARBON IMPACTS ON STRATOSPHERIC OZONE Donald J. Wuebbles Department of Atmospheric Sciences University of Illinois, Urbana, IL 61801 Tel: 217-244-1568; Fax: 217-244-4393 [email protected] INTRODUCTION Numerical models of the chemistry and physics of the global atmosphere have played a key role in the scientific understanding of past, current and potential future effects of human-related emissions of halocarbons, including Halons, other brominated compounds and various replacement compounds on stratospheric ozone and climate change. As a result, these models have led directly to the Montreal Protocol and other national and international policy decisions regarding halocarbon controls due to their effects on stratospheric ozone. The purpose of this paper is to provide a perspective on atmospheric models, the role they have played in studies of the effects of Halons and other halocarbons on ozone, and to discuss what role models will likely play in future studies of stratospheric ozone THE COMPLEXITY OF STRATOSPHERIC OZONE It is important to recognize that the stratospheric ozone layer is a naturally occurring phenomenon that has great benefits to life on Earth. In fact, the formation of the ozone layer is generally believed to have played an important role in the development of life here on Earth. The accumulation of oxygen molecules in the atmosphere allowed for the production of ozone. Gradually the increasing levels of ozone led to the formation of the stratosphere, a region of the upper atmosphere where temperature increases with altitude largely as a result of the absorption of solar radiation by ozone.