Finding Fusion Genes Resulting from Chromosome Rearrangement by Analyzing the Expressed Sequence Databases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PCDHGC3 (NM 032403) Human Tagged ORF Clone Lentiviral Particle – RC211429L4V | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC211429L4V PCDHGC3 (NM_032403) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: PCDHGC3 (NM_032403) Human Tagged ORF Clone Lentiviral Particle Symbol: PCDHGC3 Synonyms: PC43; PCDH-GAMMA-C3; PCDH2 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_032403 ORF Size: 402 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC211429). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_032403.1 RefSeq Size: 2326 bp RefSeq ORF: 405 bp Locus ID: 5098 UniProt ID: Q9BR81 Protein Families: Transmembrane MW: 13.9 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 PCDHGC3 (NM_032403) Human Tagged ORF Clone Lentiviral Particle – RC211429L4V Gene Summary: This gene is a member of the protocadherin gamma gene cluster, one of three related clusters tandemly linked on chromosome five. -

A Minimal Set of Internal Control Genes for Gene Expression Studies in Head
bioRxiv preprint doi: https://doi.org/10.1101/108381; this version posted February 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 A minimal set of internal control genes for gene expression studies in head 2 and neck squamous cell carcinoma. 1 1 1 2 3 Vinayak Palve , Manisha Pareek , Neeraja M Krishnan , Gangotri Siddappa , 2 2 1,3* 4 Amritha Suresh , Moni Abraham Kuriakose and Binay Panda 5 1 6 Ganit Labs, Bio-IT Centre, Institute of Bioinformatics and Applied Biotechnology, 7 Bangalore 560100 2 8 Mazumdar Shaw Cancer Centre and Mazumdar Shaw Centre for Translational 9 Research, Bangalore 560096 3 10 Strand Life Sciences, Bangalore 560024 11 * Corresponding author: [email protected] 12 13 Abstract: 14 Background: Selection of the right reference gene(s) is crucial in the analysis and 15 interpretation of gene expression data. In head and neck cancer, studies evaluating 16 the efficacy of internal reference genes are rare. Here, we present data for a minimal 17 set of candidates as internal control genes for gene expression studies in head and 18 neck cancer. 19 Methods: We analyzed data from multiple sources (in house whole-genome gene 20 expression microarrays, n=21; TCGA RNA-seq, n=42, and published gene 21 expression studies in head and neck tumors from literature) to come up with a set of 22 genes (discovery set) for their stable expression across tumor and normal tissues. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

PCDHGC3 (NM 032402) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC211379L3V PCDHGC3 (NM_032402) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: PCDHGC3 (NM_032402) Human Tagged ORF Clone Lentiviral Particle Symbol: PCDHGC3 Synonyms: PC43; PCDH-GAMMA-C3; PCDH2 Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) ACCN: NM_032402 ORF Size: 2589 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC211379). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_032402.1 RefSeq Size: 2794 bp RefSeq ORF: 2592 bp Locus ID: 5098 UniProt ID: Q9UN70, Q9BR81 Domains: CA Protein Families: Transmembrane MW: 91.2 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 PCDHGC3 (NM_032402) Human Tagged ORF Clone Lentiviral Particle – RC211379L3V Gene Summary: This gene is a member of the protocadherin gamma gene cluster, one of three related clusters tandemly linked on chromosome five. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Transcriptional Profiling of Breast Cancer Cells in Response to Mevinolin: Evidence of Cell Cycle Arrest, DNA Degradation and Apoptosis
1886 INTERNATIONAL JOURNAL OF ONCOLOGY 48: 1886-1894, 2016 Transcriptional profiling of breast cancer cells in response to mevinolin: Evidence of cell cycle arrest, DNA degradation and apoptosis ALI M. MAHMOUD1,2, MOURAD A.M. ABOUL-SOUD1,3, JUNKYU HAN4,5, YAZEED A. AL-SHEIKH3, AHMED M. AL-ABD6 and HANY A. EL-SHEMY1 1Department of Biochemistry, Faculty of Agriculture, Cairo University, Giza 12613; 2Centre for Aging and Associated Diseases, Helmy Institute for Medical Sciences, Zewail City of Science and Technology, Giza 12588, Egypt; 3Chair of Medical and Molecular Genetics Research, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud University, Riyadh 11433, Kingdom of Saudi Arabia; 4Graduate School of Life and Environmental Sciences; 5Alliance for Research on North Africa (ARENA), University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan; 6Department of Pharmacology, Medical Division, National Research Centre, Cairo 21622, Egypt Received January 29, 2015; Accepted March 3, 2016 DOI: 10.3892/ijo.2016.3418 Abstract. The merging of high-throughput gene expression lines. In addition, MVN‑induced transcript abundance profiles techniques, such as microarray, in the screening of natural inferred from microarrays showed significant changes in some products as anticancer agents, is considered the optimal solu- key cell processes. The changes were predicted to induce cell tion for gaining a better understanding of the intervention cycle arrest and reactive oxygen species generation but inhibit mechanism. Red yeast rice (RYR), a Chinese dietary product, DNA repair and cell proliferation. This MVN-mediated contains a mixture of hypocholesterolemia agents such as multi‑factorial stress triggered specific programmed cell death statins. -

Myopia in African Americans Is Significantly Linked to Chromosome 7P15.2-14.2
Genetics Myopia in African Americans Is Significantly Linked to Chromosome 7p15.2-14.2 Claire L. Simpson,1,2,* Anthony M. Musolf,2,* Roberto Y. Cordero,1 Jennifer B. Cordero,1 Laura Portas,2 Federico Murgia,2 Deyana D. Lewis,2 Candace D. Middlebrooks,2 Elise B. Ciner,3 Joan E. Bailey-Wilson,1,† and Dwight Stambolian4,† 1Department of Genetics, Genomics and Informatics and Department of Ophthalmology, University of Tennessee Health Science Center, Memphis, Tennessee, United States 2Computational and Statistical Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Baltimore, Maryland, United States 3The Pennsylvania College of Optometry at Salus University, Elkins Park, Pennsylvania, United States 4Department of Ophthalmology, University of Pennsylvania, Philadelphia, Pennsylvania, United States Correspondence: Joan E. PURPOSE. The purpose of this study was to perform genetic linkage analysis and associ- Bailey-Wilson, NIH/NHGRI, 333 ation analysis on exome genotyping from highly aggregated African American families Cassell Drive, Suite 1200, Baltimore, with nonpathogenic myopia. African Americans are a particularly understudied popula- MD 21131, USA; tion with respect to myopia. [email protected]. METHODS. One hundred six African American families from the Philadelphia area with a CLS and AMM contributed equally to family history of myopia were genotyped using an Illumina ExomePlus array and merged this work and should be considered co-first authors. with previous microsatellite data. Myopia was initially measured in mean spherical equiv- JEB-W and DS contributed equally alent (MSE) and converted to a binary phenotype where individuals were identified as to this work and should be affected, unaffected, or unknown. -

UNIVERSITY of CALIFORNIA RIVERSIDE Investigations Into The
UNIVERSITY OF CALIFORNIA RIVERSIDE Investigations into the Role of TAF1-mediated Phosphorylation in Gene Regulation A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Cell, Molecular and Developmental Biology by Brian James Gadd December 2012 Dissertation Committee: Dr. Xuan Liu, Chairperson Dr. Frank Sauer Dr. Frances M. Sladek Copyright by Brian James Gadd 2012 The Dissertation of Brian James Gadd is approved Committee Chairperson University of California, Riverside Acknowledgments I am thankful to Dr. Liu for her patience and support over the last eight years. I am deeply indebted to my committee members, Dr. Frank Sauer and Dr. Frances Sladek for the insightful comments on my research and this dissertation. Thanks goes out to CMDB, especially Dr. Bachant, Dr. Springer and Kathy Redd for their support. Thanks to all the members of the Liu lab both past and present. A very special thanks to the members of the Sauer lab, including Silvia, Stephane, David, Matt, Stephen, Ninuo, Toby, Josh, Alice, Alex and Flora. You have made all the years here fly by and made them so enjoyable. From the Sladek lab I want to thank Eugene, John, Linh and Karthi. Special thanks go out to all the friends I’ve made over the years here. Chris, Amber, Stephane and David, thank you so much for feeding me, encouraging me and keeping me sane. Thanks to the brothers for all your encouragement and prayers. To any I haven’t mentioned by name, I promise I haven’t forgotten all you’ve done for me during my graduate years. -

Differential Regulation of Estrogen Receptor Α Expression in Breast
Author Manuscript Published OnlineFirst on January 10, 2014; DOI: 10.1158/0008-5472.CAN-13-2020 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. CAN-13-2020R Differential Regulation of Estrogen Receptor α Expression in Breast Cancer Cells by Metastasis-Associated Protein 1 Hyun-Jin Kang1, Min-Ho Lee1, Hae-Lim Kang1, Sung-Hae Kim1, Jung-Ranh Ahn1, Hyelin Na1, Tae-Young Na1, Yona Kim2, Je Kyung Seong2, and Mi-Ock Lee1 1College of Pharmacy and Bio-MAX institute, Research Institute of Pharmaceutical Sciences; and 2College of Veterinary Medicine, BK21 Plus Program for Veterinary Science, Seoul National University, Seoul, Korea; Keywords: Estrogen receptor α; MTA1; IFI16; epigenetics, hormone sensitivity Hyun-Jin Kang, Min-Ho Lee, and Hae-Lim Kang contributed equally to this work. Corresponding Authors: Mi-Ock Lee Ph. D., College of Pharmacy, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, Korea. Phone: 82-2-880-9331; Fax: 82-2-887-2692; E-mail: [email protected] Downloaded from cancerres.aacrjournals.org on September 29, 2021. © 2014 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 10, 2014; DOI: 10.1158/0008-5472.CAN-13-2020 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. ABSTRACT Metastasis-associated protein 1 (MTA1) is a component of the nucleosome remodeling and histone deacetylase (HDAC) complex, which plays an important role in progression of breast cancer. Although MTA1 is known as a repressor of the transactivation function of estrogen receptor (ER)α, its involvement in the epigenetic control of transcription of the ERα gene ESR1 has not been studied. -
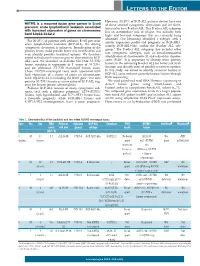
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement.