Identification of Functional Genetic Variation in Exome Sequence Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fine Mapping Studies of Quantitative Trait Loci for Baseline Platelet Count in Mice and Humans
Fine mapping studies of quantitative trait loci for baseline platelet count in mice and humans A thesis submitted in fulfillment of the requirements for the degree of Doctor of Philosophy Melody C Caramins December 2010 University of New South Wales ORIGINALITY STATEMENT ‘I hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom I have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.’ Signed …………………………………………….............. Date …………………………………………….............. This thesis is dedicated to my father. Dad, thanks for the genes – and the environment! ACKNOWLEDGEMENTS “Nothing can come out of nothing, any more than a thing can go back to nothing.” - Marcus Aurelius Antoninus A PhD thesis is never the work of one person in isolation from the world at large. I would like to thank the following people, without whom this work would not have existed. Thank you firstly, to all my teachers, of which there have been many. Undoubtedly, the greatest debt is owed to my supervisor, Dr Michael Buckley. -

MTHFR) and Methionine Synthase Reductase (MTRR
ANTICANCER RESEARCH 28 : 2807-2812 (2008) Methylenetetrahy drofolate Reductase (MTHFR) and Methionine Synthase Reductase (MTRR) Gene Polymorphisms as Risk Factors for Hepatocellular Carcinoma in a Korean Population SUN YOUNG KWAK 1,2 , UN KYUNG KIM 3, HYO JIN CHO 2, HEE KEUN LEE 3, HYE JIN KIM 2, NAM KEUN KIM 2 and SEONG GYU HWANG 1,2 1Department of Internal Medicine, 2Institute for Clinical Research, College of Medicine, Pochon CHA University, Seongnam; 3Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, South Korea Abstract. Hepatocellular carcinoma (HCC) is the third were increased risk factors for the disease. The MTHFR most frequent cause of cancer death in South Korea, but 1298A>C and the MTRR 66A>G genotypes were associated genetic susceptibility factors of HCC have not been examined with an increased risk of HCC in th is Korean population. extensively. Methylenetetrahydrofolate reductase (MTHFR) Further studies involving larger and varied populations and methionine synthase reductase (MTRR) play an essential could provide a potential tool for cancer risk assessment in role in both DNA synthesis and methylation and patients who are at risk of developing HCC. polymorphisms in the MTHFR gene, 677C>T, 1298A>C and the MTRR gene, 66A>G, are associated with several types of Hepatocellular carcinoma (HCC) is one of the most common malignancy. In this study, the allelic frequencies and malignant tumors with the third highest mortality rate among genotype distribution of three polymorphisms in the MTHFR malignancies in South Korea. According to a statistical report and MTRR genes from 96 hepatocellular carcinoma (HCC) in 2006, it occurred in 33.8/10,000 men and 11.2/10,000 patients and 201 controls were examined to assess the women in South Korea in 2005 (1). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
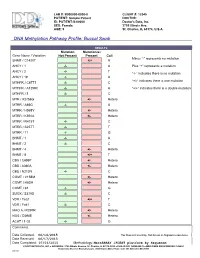
DNA Methylation Pathway Profile; Buccal Swab
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-00000 Doctor's Data, Inc. SEX: Female 3755 Illinois Ave. AGE: 5 St. Charles, IL 60174, U.S.A. !DNA Methylation Pathway Profile; Buccal Swab RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call Minus “-“ represents no mutation SHMT / C1420T +/+ A AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A “+/-“ indicates there is one mutation MTHFR / C677T -/- C MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G -/- A MTRR / H595Y +/- Hetero MTRR / K350A +/- Hetero MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 -/- G BHMT / 1 -/- A BHMT / 2 -/- C BHMT / 4 +/- Hetero BHMT / 8 +/+ T CBS / C699T +/- Hetero CBS / A360A +/- Hetero CBS / N212N -/- C COMT / V158M +/- Hetero COMT / H62H +/- Hetero COMT / 61 -/- G SUOX / S370S -/- C VDR / Taq1 +/+ T VDR / Fok1 -/- C MAO A / R297R +/- Hetero NOS / D298E +/- Hetero ACAT / 1-02 -/- G Comments: Date Collected: 06/14/2015 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 06/17/2015 Date Completed: 07/03/2015 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

Identification of Differentially Methylated Cpg
biomedicines Article Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars Mansour A. Alghamdi 1,2 , Hilary J. Wallace 3,4 , Phillip E. Melton 5,6 , Eric K. Moses 5,6, Andrew Stevenson 4 , Laith N. Al-Eitan 7,8 , Suzanne Rea 9, Janine M. Duke 4, 10 11 4,9,12 4, , Patricia L. Danielsen , Cecilia M. Prêle , Fiona M. Wood and Mark W. Fear * y 1 Department of Anatomy, College of Medicine, King Khalid University, Abha 61421, Saudi Arabia; [email protected] 2 Genomics and Personalized Medicine Unit, College of Medicine, King Khalid University, Abha 61421, Saudi Arabia 3 School of Medicine, The University of Notre Dame Australia, Fremantle 6959, Australia; [email protected] 4 Burn Injury Research Unit, School of Biomedical Sciences, Faculty of Health and Medical Sciences, The University of Western Australia, Perth 6009, Australia; andrew@fionawoodfoundation.com (A.S.); [email protected] (J.M.D.); fi[email protected] (F.M.W.) 5 Centre for Genetic Origins of Health and Disease, Faculty of Health and Medical Sciences, The University of Western Australia, Perth 6009, Australia; [email protected] (P.E.M.); [email protected] (E.K.M.) 6 School of Pharmacy and Biomedical Sciences, Faculty of Health Science, Curtin University, Perth 6102, Australia 7 Department of Applied Biological Sciences, Jordan University of Science and Technology, Irbid 22110, Jordan; [email protected] 8 Department of Biotechnology and Genetic Engineering, Jordan University of Science and Technology, Irbid 22110, -

Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network
Published OnlineFirst September 12, 2018; DOI: 10.1158/2159-8290.CD-17-1018 RESEARCH ARTICLE Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network Manon Eckhardt 1 , 2 , 3 , Wei Zhang 4 , Andrew M. Gross 4 , John Von Dollen 1 , 3 , Jeffrey R. Johnson 1 , 2 , 3 , Kathleen E. Franks-Skiba1 , 3 , Danielle L. Swaney 1 , 2 , 3 , 5 , Tasha L. Johnson 3 , Gwendolyn M. Jang 1 , 3 , Priya S. Shah1 , 3 , Toni M. Brand 6 , Jacques Archambault 7 , Jason F. Kreisberg 4 , 5 , Jennifer R. Grandis 5 , 6 , Trey Ideker4 , 5 , and Nevan J. Krogan 1 , 2 , 3 , 5 ABSTRACT We have mapped a global network of virus–host protein interactions by purifi cation of the complete set of human papillomavirus (HPV) proteins in multiple cell lines followed by mass spectrometry analysis. Integration of this map with tumor genome atlases shows that the virus targets human proteins frequently mutated in HPV − but not HPV + cancers, providing a unique opportunity to identify novel oncogenic events phenocopied by HPV infection. For example, we fi nd that the NRF2 transcriptional pathway, which protects against oxidative stress, is activated by interaction of the NRF2 regulator KEAP1 with the viral protein E1. We also demonstrate that the L2 HPV protein physically interacts with the RNF20/40 histone ubiquitination complex and promotes tumor cell inva- sion in an RNF20/40-dependent manner. This combined proteomic and genetic approach provides a systematic means to study the cellular mechanisms hijacked by virally induced cancers. SIGNIFICANCE : In this study, we created a protein–protein interaction network between HPV and human proteins. -

DNA Methylation Pathway Profile; Blood Spot
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-000000000 Doctor's Data, Inc. SEX: Male 3755 Illinois Ave AGE: 45 St. Charles, IL 60174 USA !DNA Methylation Pathway Profile; Blood Spot RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call SHMT / C1420T +/- Hetero Minus “-“ represents no mutation AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A MTHFR / C677T -/- C “+/-“ indicates there is one mutation MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G +/- Hetero MTRR / H595Y -/- C MTRR / K350A -/- A MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 +/- Hetero BHMT / 1 -/- A BHMT / 2 +/- Hetero BHMT / 4 +/+ C BHMT / 8 +/+ T CBS / C699T -/- C CBS / A360A -/- C CBS / N212N -/- C COMT / V158M +/+ A COMT / H62H +/+ T COMT / 61 -/- G SUOX / S370S -/- CG VDR / Taq1 -/- C VDR / Fok1 -/- C MAO A / R297R -/- G NOS / D298E -/- G ACAT / 1-02 +/- Hetero Comments: Date Collected: 11/08/2012 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 11/13/2012 Date Completed: 12/10/2012 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

A Master Autoantigen-Ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A Master Autoantigen-ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases Julia Y. Wang1*, Michael W. Roehrl1, Victor B. Roehrl1, and Michael H. Roehrl2* 1 Curandis, New York, USA 2 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA * Correspondence: [email protected] or [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Chronic and debilitating autoimmune sequelae pose a grave concern for the post-COVID-19 pandemic era. Based on our discovery that the glycosaminoglycan dermatan sulfate (DS) displays peculiar affinity to apoptotic cells and autoantigens (autoAgs) and that DS-autoAg complexes cooperatively stimulate autoreactive B1 cell responses, we compiled a database of 751 candidate autoAgs from six human cell types. At least 657 of these have been found to be affected by SARS-CoV-2 infection based on currently available multi-omic COVID data, and at least 400 are confirmed targets of autoantibodies in a wide array of autoimmune diseases and cancer. -

Species Specific Differences in Use of ANP32 Proteins by Influenza a Virus
Edinburgh Research Explorer Species specific differences in use of ANP32 proteins by influenza A virus Citation for published version: Long, JS, Idoko-Akoh, A, Mistry, B, Goldhill, DH, Staller , E, Schreyer, J, Ross, C, Goodbourn, S, Shelton, H, Skinner, MA, Sang, H, McGrew, M & Barclay, WS 2019, 'Species specific differences in use of ANP32 proteins by influenza A virus', eLIFE, vol. N/A, 2019;8:e45066. https://doi.org/10.7554/eLife.45066 Digital Object Identifier (DOI): 10.7554/eLife.45066 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: eLIFE Publisher Rights Statement: This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 11. Oct. 2021 -

Analysis of Seven Maternal Polymorphisms of Genes Involved In
July 2006 ⅐ Vol. 8 ⅐ No. 7 article Analysis of seven maternal polymorphisms of genes involved in homocysteine/folate metabolism and risk of Down syndrome offspring Iris Scala, MD1, Barbara Granese, PhD1, Maria Sellitto, MD1, Serena Salome`, MD1, Annalidia Sammartino, MD2, Antonio Pepe, PhD1, Pierpaolo Mastroiacovo, MD3, Gianfranco Sebastio, MD1, and Generoso Andria, MD1 PURPOSE: We present a case-control study of seven polymorphisms of six genes involved in homocysteine/folate pathway as risk factors for Down syndrome. Gene-gene/allele-allele interactions, haplotype analysis and the association with age at conception were also evaluated. METHODS: We investigated 94 Down syndrome-mothers and 264 control-women from Campania, Italy. RESULTS: Increased risk of Down syndrome was associated with the methylenetetrahydrofolate reductase (MTHFR) 1298C allele (OR 1.46; 95% CI 1.02–2.10), the MTHFR 1298CC genotype (OR 2.29; 95% CI 1.06–4.96), the reduced-folate-carrier1 (RFC1) 80G allele (1.48; 95% CI 1.05–2.10) and the RFC1 80 GG genotype (OR 2.05; 95% CI 1.03–4.07). Significant associations were found between maternal age at conception Ն34 years and either the MTHFR 1298C or the RFC 180G alleles. Positive interactions were found for the following genotype-pairs: MTHFR 677TT and 1298CC/CA, 1298CC/CA and RFC1 80 GG/GA, RFC1 80 GG and methylenetetrahydrofolate-dehydrogenase 1958 AA. The 677–1298 T-C haplotype at the MTHFR locus was also a risk factor for Down syndrome (P ϭ 0.0022). The methionine-synthase-reductase A66G, the methionine-synthase A2756G and the cystathionine-beta-synthase 844ins68 polymorphisms were not associated with increased risk of Down syndrome. -

To See an Example Report
John Doe 02/16/2016 Recommendations based on DNA results – Please follow only under the advice and care of your physician. • Minimize caffeine • Avoid ibuprofen and naproxen regularly and you may need reduced doses of blood thinners if you ever need them • See handout on CYP2D6 for possible drug sensitivities • Avoid GMO foods and high oxalate foods if have been using antibiotics or gut needs healing • Avoid gluten containing foods and supplements. Also consider eliminating dairy and soy • If cholesterol high, watch cholesterol in foods • Check RBC folate and homocysteine levels • Check ammonia and molybdenum levels • Avoid fluoroquinolones, such as Cipro and Levaquin • Eat beets, lamb, and spinach as a source of betaine • Do you have chemical sensitivities to perfume or cigarette smoke? (possible need for SAMe or phosphatidyl choline) • Do you experience anxiety or panic attacks? (betaine, TMG, check RBC folate levels) • If sensitivities to sulfur foods, remove for three days, then introduce back in and see how you feel. Ammonia and molybdenum levels can also be checked for a block in sulfur pathway. If sulfur a problem, stay away from oxalate foods and address possible leaky gut. Supplement Recommendations • Nattoserrazimes (CYP4V2, ITGB3, NR1I12, F11) • Phosphatidyl choline (PEMT; if multiple chemical sensitivities) • Molybdenum (SUOX; check multi vitamin) • Sam-e (AHCY, MTR/MTRR) • TMG (BHMT, but check for anxiety because of COMT) • Quatrefolic (5-MTHF; MTHFR, MTHFD1, MTHFR, MTHFS, MTR/MTRR, PEMT, MAO A) • Probiotic synergy (FUT2) • 5-HTP (MAO A) • PharmaGABA (GAD1) • Adenosyl B12 (COMT, TCN1/2) • Cod liver oil (for vitamin A; BCMO1) • Mitochondrial NRG (NDUFS7) © Ruthieharper MD What You Need to Know In the world of genetic testing, things can become complicated very quickly. -

Human Leucine-Rich Repeat Proteins: a Genome-Wide Bioinformatic Categorization and Functional Analysis in Innate Immunity
Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity Aylwin C. Y. Nga,b,1, Jason M. Eisenberga,b,1, Robert J. W. Heatha, Alan Huetta, Cory M. Robinsonc, Gerard J. Nauc, and Ramnik J. Xaviera,b,2 aCenter for Computational and Integrative Biology, and Gastrointestinal Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114; bThe Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; and cMicrobiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261 Edited by Jeffrey I. Gordon, Washington University School of Medicine, St. Louis, MO, and approved June 11, 2010 (received for review February 17, 2010) In innate immune sensing, the detection of pathogen-associated proteins have been implicated in human diseases to date, notably molecular patterns by recognition receptors typically involve polymorphisms in NOD2 in Crohn disease (8, 9), CIITA in leucine-rich repeats (LRRs). We provide a categorization of 375 rheumatoid arthritis and multiple sclerosis (10), and TLR5 in human LRR-containing proteins, almost half of which lack other Legionnaire disease (11). identifiable functional domains. We clustered human LRR proteins Most LRR domains consist of a chain of between 2 and 45 by first assigning LRRs to LRR classes and then grouping the proteins LRRs (12). Each repeat in turn is typically 20 to 30 residues long based on these class assignments, revealing several of the resulting and can be divided into a highly conserved segment (HCS) fol- protein groups containing a large number of proteins with certain lowed by a variable segment (VS).