Chromatin Control of Viral Infection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials: Evaluation of Cytotoxicity and Α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids
Supplementary Materials: Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids Oxana B. Kazakova1*, Gul'nara V. Giniyatullina1, Akhat G. Mustafin1, Denis A. Babkov2, Elena V. Sokolova2, Alexander A. Spasov2* 1Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences, 71, pr. Oktyabrya, 450054 Ufa, Russian Federation 2Scientific Center for Innovative Drugs, Volgograd State Medical University, Novorossiyskaya st. 39, Volgograd 400087, Russian Federation Correspondence Prof. Dr. Oxana B. Kazakova Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences 71 Prospeсt Oktyabrya Ufa, 450054 Russian Federation E-mail: [email protected] Prof. Dr. Alexander A. Spasov Scientific Center for Innovative Drugs of the Volgograd State Medical University 39 Novorossiyskaya st. Volgograd, 400087 Russian Federation E-mail: [email protected] Figure S1. 1H and 13C of compound 2. H NH N H O H O H 2 2 Figure S2. 1H and 13C of compound 4. NH2 O H O H CH3 O O H H3C O H 4 3 Figure S3. Anticancer screening data of compound 2 at single dose assay 4 Figure S4. Anticancer screening data of compound 7 at single dose assay 5 Figure S5. Anticancer screening data of compound 8 at single dose assay 6 Figure S6. Anticancer screening data of compound 9 at single dose assay 7 Figure S7. Anticancer screening data of compound 12 at single dose assay 8 Figure S8. Anticancer screening data of compound 13 at single dose assay 9 Figure S9. Anticancer screening data of compound 14 at single dose assay 10 Figure S10. -

Functional Compensation Among HMGN Variants Modulates the Dnase I Hypersensitive Sites at Enhancers
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Research Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers Tao Deng,1,12 Z. Iris Zhu,2,12 Shaofei Zhang,1 Yuri Postnikov,1 Di Huang,2 Marion Horsch,3 Takashi Furusawa,1 Johannes Beckers,3,4,5 Jan Rozman,3,5 Martin Klingenspor,6,7 Oana Amarie,3,8 Jochen Graw,3,8 Birgit Rathkolb,3,5,9 Eckhard Wolf,9 Thure Adler,3 Dirk H. Busch,10 Valérie Gailus-Durner,3 Helmut Fuchs,3 Martin Hrabeˇ de Angelis,3,4,5 Arjan van der Velde,2,13 Lino Tessarollo,11 Ivan Ovcherenko,2 David Landsman,2 and Michael Bustin1 1Protein Section, Laboratory of Metabolism, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA; 2Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20892, USA; 3German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 4Experimental Genetics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, 85354 Freising-Weihenstephan, Germany; 5German Center for Diabetes Research (DZD), 85764 Neuherberg, Germany; 6Molecular Nutritional Medicine, Technische Universität München, 85350 Freising, Germany; 7Center for Nutrition and Food Sciences, Technische Universität München, 85350 Freising, Germany; 8Institute of Developmental Genetics (IDG), 85764 -

Changes to Virus Taxonomy 2004
Arch Virol (2005) 150: 189–198 DOI 10.1007/s00705-004-0429-1 Changes to virus taxonomy 2004 M. A. Mayo (ICTV Secretary) Scottish Crop Research Institute, Invergowrie, Dundee, U.K. Received July 30, 2004; accepted September 25, 2004 Published online November 10, 2004 c Springer-Verlag 2004 This note presents a compilation of recent changes to virus taxonomy decided by voting by the ICTV membership following recommendations from the ICTV Executive Committee. The changes are presented in the Table as decisions promoted by the Subcommittees of the EC and are grouped according to the major hosts of the viruses involved. These new taxa will be presented in more detail in the 8th ICTV Report scheduled to be published near the end of 2004 (Fauquet et al., 2004). Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., and Ball, L.A. (eds) (2004). Virus Taxonomy, VIIIth Report of the ICTV. Elsevier/Academic Press, London, pp. 1258. Recent changes to virus taxonomy Viruses of vertebrates Family Arenaviridae • Designate Cupixi virus as a species in the genus Arenavirus • Designate Bear Canyon virus as a species in the genus Arenavirus • Designate Allpahuayo virus as a species in the genus Arenavirus Family Birnaviridae • Assign Blotched snakehead virus as an unassigned species in family Birnaviridae Family Circoviridae • Create a new genus (Anellovirus) with Torque teno virus as type species Family Coronaviridae • Recognize a new species Severe acute respiratory syndrome coronavirus in the genus Coro- navirus, family Coronaviridae, order Nidovirales -

Examination of the Transcription Factors Acting in Bone Marrow
THESIS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY (PHD) Examination of the transcription factors acting in bone marrow derived macrophages by Gergely Nagy Supervisor: Dr. Endre Barta UNIVERSITY OF DEBRECEN DOCTORAL SCHOOL OF MOLECULAR CELL AND IMMUNE BIOLOGY DEBRECEN, 2016 Table of contents Table of contents ........................................................................................................................ 2 1. Introduction ............................................................................................................................ 5 1.1. Transcriptional regulation ................................................................................................... 5 1.1.1. Transcriptional initiation .................................................................................................. 5 1.1.2. Co-regulators and histone modifications .......................................................................... 8 1.2. Promoter and enhancer sequences guiding transcription factors ...................................... 11 1.2.1. General transcription factors .......................................................................................... 11 1.2.2. The ETS superfamily ..................................................................................................... 17 1.2.3. The AP-1 and CREB proteins ........................................................................................ 20 1.2.4. Other promoter specific transcription factor families ................................................... -

HMGB1 in Health and Disease R
Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2014 HMGB1 in health and disease R. Kang R. C. Chen Q. H. Zhang W. Hou S. Wu See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Emergency Medicine Commons Recommended Citation Kang R, Chen R, Zhang Q, Hou W, Wu S, Fan X, Yan Z, Sun X, Wang H, Tang D, . HMGB1 in health and disease. 2014 Jan 01; 40():Article 533 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/533. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors R. Kang, R. C. Chen, Q. H. Zhang, W. Hou, S. Wu, X. G. Fan, Z. W. Yan, X. F. Sun, H. C. Wang, D. L. Tang, and +8 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/533 NIH Public Access Author Manuscript Mol Aspects Med. Author manuscript; available in PMC 2015 December 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Aspects Med. 2014 December ; 0: 1–116. doi:10.1016/j.mam.2014.05.001. HMGB1 in Health and Disease Rui Kang1,*, Ruochan Chen1, Qiuhong Zhang1, Wen Hou1, Sha Wu1, Lizhi Cao2, Jin Huang3, Yan Yu2, Xue-gong Fan4, Zhengwen Yan1,5, Xiaofang Sun6, Haichao Wang7, Qingde Wang1, Allan Tsung1, Timothy R. -

Hepatitis C Virus As a Unique Human Model Disease to Define
viruses Review Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection David Wolski and Georg M. Lauer * Liver Center at the Gastrointestinal Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA * Correspondence: [email protected]; Tel.: +1-617-724-7515 Received: 27 June 2019; Accepted: 23 July 2019; Published: 26 July 2019 Abstract: The hepatitis C virus is unique among chronic viral infections in that an acute outcome with complete viral elimination is observed in a minority of infected patients. This unique feature allows direct comparison of successful immune responses with those that fail in the setting of the same human infection. Here we review how this scenario can be used to achieve better understanding of transcriptional regulation of T-cell differentiation. Specifically, we discuss results from a study comparing transcriptional profiles of hepatitis C virus (HCV)-specific CD8 T-cells during early HCV infection between patients that do and do not control and eliminate HCV. Identification of early gene expression differences in key T-cell differentiation molecules as well as clearly distinct transcriptional networks related to cell metabolism and nucleosomal regulation reveal novel insights into the development of exhausted and memory T-cells. With additional transcriptional studies of HCV-specific CD4 and CD8 T-cells in different stages of infection currently underway, we expect HCV infection to become a valuable model disease to study human immunity to viruses. Keywords: viral hepatitis; hepatitis C virus; T cells; transcriptional regulation; transcription factors; metabolism; nucleosome 1. -

Eric Verdin, MD
CURRICULUM VITAE Eric Verdin, MD Work Address The Buck Institute for Research on Aging 8001 Redwood Blvd. Novato, CA 94945 Office Phone: (415) 209-2250 Cell Phone: (415) 305-9208 Email: [email protected] Biographical Data Birthplace: Belgium Citizenship: Belgium, USA Education MD, University of Liège Liège, Belgium, 1982 Visiting Student, Harvard Medical School Boston, MA, 1981 B.S. in Medical Sciences, University of Liège Liège, Belgium, 1978 Research Positions 2016- Present CEO & President, The Buck Institute for Research on Aging, Novato, CA 2016- Present Adjunct Professor, Leonard Davis School of Gerontology, University of Southern California, Los Angeles 2004-2016 Associate Director, Gladstone Institutes, San Francisco, CA 1998- Present Professor, University of California, San Francisco, CA 1997- 2016 Senior Investigator, Gladstone Institute, San Francisco, CA 1997 Professor, The Picower Institute for Medical Research, Manhasset, NY 1993-1997 Associate Professor, The Picower Institute for Medical Research, Manhasset, NY 1990-1993 Senior Staff Fellow, NIH, Bethesda, MD 1 1987-1990 Assistant Professor, Free University of Brussels, Belgium 1984-1987 Postdoctoral Fellowship, Harvard Medical School, Elliott P. Joslin Research Laboratory, Boston, MA (Laboratories of C. Ronald Kahn and Bernard Fields) 1983-1984 Intern in Medicine, University of Massachusetts, Worcester, MA 1982-1983 Research Intern, University of Liège, Department of Internal Medicine, Liège, Belgium Memberships & Activities Member, Board of Directors, Bay Area -

Combinatorial Bzip Dimers Display Complex DNA-Binding Specificity Landscapes
Combinatorial bZIP dimers display complex DNA-binding specificity landscapes The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rodriguez-Martinez, Jose A et al. “Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes.” eLife 6 (2017): n. pag. As Published http://dx.doi.org/10.7554/eLife.19272 Publisher eLife Sciences Publications, Ltd. Version Final published version Citable link http://hdl.handle.net/1721.1/110147 Terms of Use Creative Commons Attribution 4.0 International License Detailed Terms http://creativecommons.org/licenses/by-nc/4.0/ RESEARCH ARTICLE Combinatorial bZIP dimers display complex DNA-binding specificity landscapes Jose´ A Rodrı´guez-Martı´nez1†, Aaron W Reinke2†, Devesh Bhimsaria1,3†, Amy E Keating2,4, Aseem Z Ansari1,5* 1Department of Biochemistry, University of Wisconsin-Madison, Madison, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Electrical and Computer Engineering, University of Wisconsin-Madison, Madison, Unites States; 4Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, United States; 5The Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, United States Abstract How transcription factor dimerization impacts DNA-binding specificity is poorly understood. Guided by protein dimerization properties, we examined DNA binding specificities of 270 human bZIP pairs. DNA interactomes of 80 heterodimers and 22 homodimers revealed that 72% of heterodimer motifs correspond to conjoined half-sites preferred by partnering monomers. Remarkably, the remaining motifs are composed of variably-spaced half-sites (12%) or ‘emergent’ sites (16%) that cannot be readily inferred from half-site preferences of partnering monomers. -

CRISPR-Based COVID-19 Smartphone Test in Development
News & Analysis Biotech Innovations CRISPR-Based COVID-19 Smartphone Predicting COVID-19 Outcomes any symptoms, to make sure resources are Test in Development in Emergency Department Patients appropriately allocated,” Fred Kwon, PhD, A simplified point-of-care assay that turns a A new artificial intelligence algorithm uses the study’s lead author, said in a statement. smartphone into a fluorescence micro- chest x-ray severity scores and clinical vari- The chest x-ray severity scores alone could scope could expand coronavirus disease ables collected during emergency depart- be useful. “We are working to incorporate 2019(COVID-19)testingcapability,research- ment (ED) visits to predict whether pa- this algorithm-generated severity score into ers reported in a study in Cell. tients with coronavirus disease 2019 the clinical workflow to inform treatment de- The assay, which uses clustered (COVID-19) will be intubated or will die. If fur- cisions and flag high-risk patients in the fu- regularly interspaced short palindromic ther validated in larger studies with addi- ture,” Kwon said. repeats (CRISPR) gene editing technol- tional patient populations, the proof-of- ogy, emits a fluo- concept model, described in Radiology: Phone Apps and Wearable Trackers rescent signal in Artificial Intelligence, could be used to ap- Modestly Improve Activity Viewpoint page 529 the presence of propriately triage ED patients before they Smartphone fitness applications and wear- the novel corona- become seriously ill with COVID-19. able activity trackers seem to have a small to virus’s RNA. A smartphone camera can moderate positive effect on physical activ- detect this signal directly, without amplifi- ity,a systematic review and meta-analysis of cation of the viral genome used in most randomized trials found. -
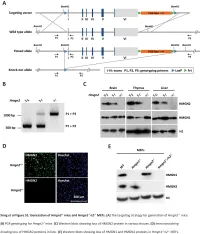
Supplemental Data.Pdf
Table S1. Summary of sequencing results A. DNase‐seq data % Align % Mismatch % >=Q30 bases Sample ID # Reads % unique reads (PF) Rate (PF) (PF) WT_1 84,301,522 86.76 0.52 93.34 96.30 WT_2 98,744,222 84.97 0.51 93.75 89.94 Hmgn1‐/‐_1 79,620,656 83.94 0.86 90.58 88.65 Hmgn1‐/‐_2 62,673,782 84.13 0.87 91.30 89.18 Hmgn2‐/‐_1 87,734,440 83.49 0.71 91.81 90.00 Hmgn2‐/‐_2 82,498,808 83.25 0.69 92.73 90.66 Hmgn1‐/‐n2‐/‐_1 71,739,638 68.51 2.31 81.11 89.22 Hmgn1‐/‐n2‐/‐_2 74,113,682 68.19 2.37 81.16 86.57 B. ChIP‐seq data Histone % Align % Mismatch % >=Q30 % unique Genotypes # Reads marks (PF) Rate (PF) bases (PF) reads H3K4me1 100670054 92.99 0.28 91.21 87.29 H3K4me3 67064272 91.97 0.35 89.11 27.15 WT H3K27ac 90,340,242 93.57 0.28 95.02 89.80 input 111,292,572 78.24 0.55 96.07 86.99 H3K4me1 84598176 92.34 0.33 91.2 81.69 H3K4me3 90032064 92.19 0.44 88.76 15.81 Hmgn1‐/‐n2‐/‐ H3K27ac 86,260,526 93.40 0.29 94.94 87.49 input 78,142,334 78.47 0.56 95.82 81.11 C. MNase‐seq data % Mismatch % >=Q30 bases % unique Sample ID # Reads % Align (PF) Rate (PF) (PF) reads WT_1_Extensive 45,232,694 55.23 1.49 90.22 81.73 WT_1_Limited 105,460,950 58.03 1.39 90.81 79.62 WT_2_Extensive 40,785,338 67.34 1.06 89.76 89.60 WT_2_Limited 105,738,078 68.34 1.05 90.29 85.96 Hmgn1‐/‐n2‐/‐_1_Extensive 117,927,050 55.74 1.49 89.50 78.01 Hmgn1‐/‐n2‐/‐_1_Limited 61,846,742 63.76 1.22 90.57 84.55 Hmgn1‐/‐n2‐/‐_2_Extensive 137,673,830 60.04 1.30 89.28 78.99 Hmgn1‐/‐n2‐/‐_2_Limited 45,696,614 62.70 1.21 90.71 85.52 D. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

ICTV Code Assigned: 2011.001Ag Officers)
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the separate document “Help with completing a taxonomic proposal” Please try to keep related proposals within a single document; you can copy the modules to create more than one genus within a new family, for example. MODULE 1: TITLE, AUTHORS, etc (to be completed by ICTV Code assigned: 2011.001aG officers) Short title: Change existing virus species names to non-Latinized binomials (e.g. 6 new species in the genus Zetavirus) Modules attached 1 2 3 4 5 (modules 1 and 9 are required) 6 7 8 9 Author(s) with e-mail address(es) of the proposer: Van Regenmortel Marc, [email protected] Burke Donald, [email protected] Calisher Charles, [email protected] Dietzgen Ralf, [email protected] Fauquet Claude, [email protected] Ghabrial Said, [email protected] Jahrling Peter, [email protected] Johnson Karl, [email protected] Holbrook Michael, [email protected] Horzinek Marian, [email protected] Keil Guenther, [email protected] Kuhn Jens, [email protected] Mahy Brian, [email protected] Martelli Giovanni, [email protected] Pringle Craig, [email protected] Rybicki Ed, [email protected] Skern Tim, [email protected] Tesh Robert, [email protected] Wahl-Jensen Victoria, [email protected] Walker Peter, [email protected] Weaver Scott, [email protected] List the ICTV study group(s) that have seen this proposal: A list of study groups and contacts is provided at http://www.ictvonline.org/subcommittees.asp .