Ch. 1 Intro and Review 1.1 Intro to Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Electronic Structures and Spin Topologies of #-Picoliniumyl Radicals
Subscriber access provided by UNIV OF MISSOURI COLUMBIA Article Electronic Structures and Spin Topologies of #-Picoliniumyl Radicals. A Study of the Homolysis of N-Methyl-#-picolinium and of Benzo-, Dibenzo-, and Naphthoannulated Analogs Rainer Glaser, Yongqiang Sui, Ujjal Sarkar, and Kent S. Gates J. Phys. Chem. A, 2008, 112 (21), 4800-4814 • DOI: 10.1021/jp8011987 • Publication Date (Web): 22 May 2008 Downloaded from http://pubs.acs.org on December 12, 2008 More About This Article Additional resources and features associated with this article are available within the HTML version: • Supporting Information • Access to high resolution figures • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article The Journal of Physical Chemistry A is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 4800 J. Phys. Chem. A 2008, 112, 4800–4814 Electronic Structures and Spin Topologies of γ-Picoliniumyl Radicals. A Study of the Homolysis of N-Methyl-γ-picolinium and of Benzo-, Dibenzo-, and Naphthoannulated Analogs Rainer Glaser,*,† Yongqiang Sui,† Ujjal Sarkar,† and Kent S. Gates*,†,‡ Departments of Chemistry and Biochemistry, UniVersity of Missouri-Columbia, Columbia, Missouri 65211 ReceiVed: February 9, 2008 Radicals resulting from one-electron reduction of (N-methylpyridinium-4-yl) methyl esters have been reported to yield (N-methylpyridinium-4-yl) methyl radical, or N-methyl-γ-picoliniumyl for short, by heterolytic cleavage of carboxylate. This new reaction could provide the foundation for a new structural class of bioreductively activated, hypoxia-selective antitumor agents. N-methyl-γ-picoliniumyl radicals are likely to damage DNA by way of H-abstraction and it is of paramount significance to assess their H-abstraction capabilities. -

An Indicator of Triplet State Baird-Aromaticity
inorganics Article The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub 1,2, Kjell Jorner 1,2 ID and Henrik Ottosson 1,2,* 1 Department of Chemistry—BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden; [email protected] (R.A.); [email protected] (K.J.) 2 Department of Chemistry-Ångström Laboratory Uppsala University, Box 523, SE-751 20 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-18-4717476 Received: 23 October 2017; Accepted: 11 December 2017; Published: 15 December 2017 Abstract: Baird’s rule tells that the electron counts for aromaticity and antiaromaticity in the first ππ* triplet and singlet excited states (T1 and S1) are opposite to those in the ground state (S0). Our hypothesis is that a silacyclobutene (SCB) ring fused with a [4n]annulene will remain closed in the T1 state so as to retain T1 aromaticity of the annulene while it will ring-open when fused to a [4n + 2]annulene in order to alleviate T1 antiaromaticity. This feature should allow the SCB ring to function as an indicator for triplet state aromaticity. Quantum chemical calculations of energy and (anti)aromaticity changes along the reaction paths in the T1 state support our hypothesis. The SCB ring should indicate T1 aromaticity of [4n]annulenes by being photoinert except when fused to cyclobutadiene, where it ring-opens due to ring-strain relief. Keywords: Baird’s rule; computational chemistry; excited state aromaticity; Photostability 1. Introduction Baird showed in 1972 that the rules for aromaticity and antiaromaticity of annulenes are reversed in the lowest ππ* triplet state (T1) when compared to Hückel’s rule for the electronic ground state (S0)[1–3]. -

Polyphenolic Compounds Extracted and Purified from Buddleja Globosa
molecules Article Polyphenolic Compounds Extracted and Purified from Buddleja Globosa Hope (Buddlejaceae) Leaves Using Natural Deep Eutectic Solvents and Centrifugal Partition Chromatography Jeniffer Torres-Vega 1 , Sergio Gómez-Alonso 2 , José Pérez-Navarro 2 , Julio Alarcón-Enos 3 and Edgar Pastene-Navarrete 1,3,* 1 Laboratorio de Farmacognosia, Departamento de Farmacia, Facultad de Farmacia, Universidad de Concepción, Concepción PC4030000, Chile; [email protected] 2 Regional Institute for Applied Scientific Research, Faculty of Chemical Sciences, University of Castilla-La Mancha, PC13071 Castilla-La Mancha, Spain; [email protected] (S.G.-A.); [email protected] (J.P.-N.) 3 Laboratorio de Síntesis y Biotransformación de Productos Naturales, Universidad del Bío-Bío, Chillán PC3800708, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-(42)-246-3000 Abstract: Chemical profiling of Buddleja globosa was performed by high-performance liquid chro- matography coupled to electrospray ionization (HPLC-DAD-ESI-IT/MS) and quadrupole time-of-flight high-resolution mass spectrometry (HPLC-ESI-QTOF/MS). The identification of 17 main phenolic com- pounds in B. globosa leaf extracts was achieved. Along with caffeoyl glucoside isomers, caffeoylshikimic Citation: Torres-Vega, J.; acid and several verbascoside derivatives (β-hydroxyverbascoside and β-hydroxyisoverbascoside) were Gómez-Alonso, S.; Pérez-Navarro, J.; Alarcón-Enos, J.; Pastene-Navarrete, identified. Among flavonoid compounds, the presence of 6-hydroxyluteolin-7-O-glucoside, quercetin-3- E. Polyphenolic Compounds O-glucoside, luteolin 7-O-glucoside, apigenin 7-O-glucoside was confirmed. Campneoside I, forsytho- Extracted and Purified from Buddleja side B, lipedoside A and forsythoside A were identified along with verbascoside, isoverbascoside, Globosa Hope (Buddlejaceae) Leaves eukovoside and martynoside. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -
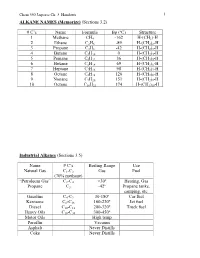
C's Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)
Chem 350 Jasperse Ch. 3 Handouts 1 ALKANE NAMES (Memorize) (Sections 3.2) # C’s Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)-H 2 Ethane C2H6 -89 H-(CH2)2-H 3 Propane C3H8 -42 H-(CH2)3-H 4 Butane C4H10 0 H-(CH2)4-H 5 Pentane C5H12 36 H-(CH2)5-H 6 Hexane C6H14 69 H-(CH2)6-H 7 Heptane C7H16 98 H-(CH2)7-H 8 Octane C8H18 126 H-(CH2)8-H 9 Nonane C9H20 151 H-(CH2)9-H 10 Octane C10H22 174 H-(CH2)10-H Industrial Alkanes (Sections 3.5) Name # C’s Boiling Range Use Natural Gas C1-C3 Gas Fuel (70% methane) “Petroleum Gas” C2-C4 <30º Heating, Gas Propane C3 -42º Propane tanks, camping, etc. Gasoline C4-C9 30-180º Car fuel Kerosene C8-C16 160-230º Jet fuel Diesel C10-C18 200-320º Truck fuel Heavy Oils C16-C30 300-450º Motor Oils High temp Paraffin Vacuum Asphalt Never Distills Coke Never Distills Chem 350 Jasperse Ch. 3 Handouts 2 Nomenclature of Alkanes (Sections 3.3) Systematic IUPAC Rules for Branched and Substituted Alkanes (Section 3.3B) 1. Longest continuous C-chain “core name” 2. Number core chain from an end nearest a substituent 3. Name substituents as “alkyl” groups: 4. Specify the location of substituents using numbers (hyphenate the #’s) • If >2 substituents, list alphabetically • Use di-, tri-, tetra- if the same substituent is repeated. (But ignore these in alphabetizing). Punctuation Notes: • Hyphenate numbers • Do not put a space between substituents and the core name Special Names for Some 3 or 4-carbon Substituents H3C CH3 CH Memorize H3C C H3C CH3 Isopropyl t-butyl or tert-butyl H2 H2 CH3 H3C C H3C C CH H3C CH Others C C C C CH3 H3C C H2 H H H2 2 2 H2 n-propyl n-butyl isobutyl s-butyl (n for "normal") Another Classification System Primary (1º): with one attached carbon Secondary (2º): with two attached carbons Tertiary (3º): with three attached carbons H C C C C C C C C H 1º H 2º C 3º Very Complex Substituents (Not responsible) Substituent: (1-ethyl-2,3-dimethylpentyl) Overall: 9-(1-ethyl-2,3-dimethylpentyl)nonadecane Chem 350 Jasperse Ch. -

Influence of Conjugation Axis on the Optical and Electronic Properties Of
Article pubs.acs.org/joc Influence of Conjugation Axis on the Optical and Electronic Properties of Aryl-Substituted Benzobisoxazoles † ‡ ‡ † † Brian C. Tlach, Aimeé L. Tomlinson, Alden G. Ryno, Dawn D. Knoble, Dana L. Drochner, † Kyle J. Krager, and Malika Jeffries-EL*, † Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States ‡ Department of Chemistry, University of North Georgia, Dahlonega, Georgia 30597, United States *S Supporting Information ABSTRACT: Six different 2,6-diethyl-4,8-diarylbenzo[1,2-d:4,5-d′]bis(oxazoles) and four different 2,4,6,8-tetraarylbenzobisoxazoles were synthesized in two steps: a Lewis acid catalyzed orthoester cyclization followed by a Suzuki or Stille cross-coupling with various arenes. The influence of aryl group substitution and/or conjugation axis variation on the optical and electronic properties of these benzobis(oxazole) (BBO) compounds was evaluated. Structural modifications could be used to alter the HOMO, LUMO, and band gap over a range of 1.0, 0.5, and 0.5 eV, respectively. However, depending on the location and identity of the substituent, the HOMO level can be altered without significantly impacting the LUMO level. This is supported by the calculated frontier molecular orbitals. Our results indicate that the FMOs and band gaps of benzobisoxazoles can be readily modified either jointly or individually. ■ INTRODUCTION Among the aforementioned examples, the benzo[1,2-d:4,5- d′]bis(oxazole) (BBO)-based cruciforms are particularly During the past four decades, interest in the development of π- interesting, since these molecules have two different con- conjugated materials has increased due to their potential use as jugation pathways: 2,6-conjugation through the oxazole rings replacements for inorganic materials in a variety of semi- and 4,8-conjugation through the central benzene ring (Scheme conducting applications including field effect transistors − − 1). -

Mechanistic Approach to Thermal Production of New Materials from Asphaltenes of Castilla Crude Oil
processes Article Mechanistic Approach to Thermal Production of New Materials from Asphaltenes of Castilla Crude Oil Natalia Afanasjeva 1, Andrea González-Córdoba 2,* and Manuel Palencia 1 1 Department of Chemistry, Faculty of Natural and Exact Science, Universidad del Valle, Cali 760020, Colombia; [email protected] (N.A.); [email protected] (M.P.) 2 Department of Chemical and Environmental Engineering, Faculty of Engineering, Universidad Nacional de Colombia, Bogotá 111321, Colombia * Correspondence: [email protected]; Tel.: +57-1-316-5000 (ext. 14322) Received: 16 August 2020; Accepted: 23 October 2020; Published: 12 December 2020 Abstract: Asphaltenes are compounds present in crude oils that influence their rheology, raising problems related to the extraction, transport, and refining. This work centered on the chemical and structural changes of the asphaltenes from the heavy Colombian Castilla crude oil during pyrolysis between 330 and 450 ◦C. Also, the development of new strategies to apply these macromolecules, and the possible use of the cracking products as a source of new materials were analyzed. The obtained products (coke, liquid, and gas) were collected and evaluated through the techniques of proton and carbon-13 nuclear magnetic resonance (1H and 13C NMR), elemental composition, Fourier-transform infrared spectroscopy (FTIR), X-ray powder diffraction (XRD), saturates, aromatics, resins, and asphaltenes (SARA) analysis, and gas chromatography–mass spectrometry (GC-MS). A comparison of the applied methods showed that the asphaltene molecules increased the average size of their aromatic sheets, lost their aliphatic chains, condensed their aromatic groups, and increased their degree of unsaturation during pyrolysis. In the liquid products were identified alkylbenzenes, n-alkanes C9–C30, and n-alkenes. -

Developing a S Ystem to Study the Dynamics of the Heterolysis of Psubstituted Radicals in Terms of Magnetic Field Effects
Developing a S ystem to Study the Dynamics of the Heterolysis of PSubstituted Radicals in terms of Magnetic Field Effects by Elaine K. Adams Submitted in partial fulfiiIlment of the requirements for the degree of Master of Science Dalhousie University Halifax, Nova Scotia September, 1998 @ Copyright by Elaine K. Adams, 1998 National hirary Bibliothèque nationale du Canada Acquisitions and Acquisiins et Biliograpfii Services seMces bibliographiques The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or seli reproduire, prêter, distriiuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de micro fi ch el^ de reproduction sur papier ou sur fonnat électronique. The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fkom it Ni la thése ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation. Table of Contents List of Figures ........................................................................................ vi ... List of Tables ........................................................................................ xm 1.1 General Introduction ....................................................................... -

Coordinate Covalent C F B Bonding in Phenylborates and Latent Formation of Phenyl Anions from Phenylboronic Acid†
J. Phys. Chem. A 2006, 110, 1295-1304 1295 Coordinate Covalent C f B Bonding in Phenylborates and Latent Formation of Phenyl Anions from Phenylboronic Acid† Rainer Glaser* and Nathan Knotts Department of Chemistry, UniVersity of MissourisColumbia, Columbia, Missouri 65211 ReceiVed: July 4, 2005; In Final Form: August 8, 2005 The results are reported of a theoretical study of the addition of small nucleophiles Nu- (HO-,F-)to - phenylboronic acid Ph-B(OH)2 and of the stability of the resulting complexes [Ph-B(OH)2Nu] with regard - - - - - to Ph-B heterolysis [Ph-B(OH)2Nu] f Ph + B(OH)2Nu as well as Nu /Ph substitution [Ph-B(OH)2Nu] - - - + Nu f Ph + [B(OH)2Nu2] . These reactions are of fundamental importance for the Suzuki-Miyaura cross-coupling reaction and many other processes in chemistry and biology that involve phenylboronic acids. The species were characterized by potential energy surface analysis (B3LYP/6-31+G*), examined by electronic structure analysis (B3LYP/6-311++G**), and reaction energies (CCSD/6-311++G**) and solvation energies - (PCM and IPCM, B3LYP/6-311++G**) were determined. It is shown that Ph-B bonding in [Ph-B(OH)2Nu] is coordinate covalent and rather weak (<50 kcal‚mol-1). The coordinate covalent bonding is large enough to inhibit unimolecular dissociation and bimolecular nucleophile-assisted phenyl anion liberation is slowed greatly by the negative charge on the borate’s periphery. The latter is the major reason for the extraordinary differences in the kinetic stabilities of diazonium ions and borates in nucleophilic substitution reactions despite their rather similar coordinate covalent bond strengths. -

Hydrogen Peroxide As a Hydride Donor and Reductant Under Biologically Relevant Conditions† Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Hydrogen peroxide as a hydride donor and reductant under biologically relevant conditions† Cite this: Chem. Sci.,2019,10,2025 ab d c All publication charges for this article Yamin Htet, Zhuomin Lu, Sunia A. Trauger have been paid for by the Royal Society and Andrew G. Tennyson *def of Chemistry Some ruthenium–hydride complexes react with O2 to yield H2O2, therefore the principle of microscopic À reversibility dictates that the reverse reaction is also possible, that H2O2 could transfer an H to a Ru complex. Mechanistic evidence is presented, using the Ru-catalyzed ABTScÀ reduction reaction as a probe, which suggests that a Ru–H intermediate is formed via deinsertion of O2 from H2O2 following Received 5th December 2018 À coordination to Ru. This demonstration that H O can function as an H donor and reductant under Accepted 7th December 2018 2 2 biologically-relevant conditions provides the proof-of-concept that H2O2 may function as a reductant in DOI: 10.1039/c8sc05418e living systems, ranging from metalloenzyme-catalyzed reactions to cellular redox homeostasis, and that À rsc.li/chemical-science H2O2 may be viable as an environmentally-friendly reductant and H source in green catalysis. Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Introduction bond and be subsequently released as H2O2 (Scheme 1A, red arrows).12,13 The principle of microscopic reversibility14 there- Hydrogen peroxide and its descendant reactive oxygen species fore dictates that it is mechanistically equivalent for H2O2 to (ROS) have historically been viewed in biological systems nearly react with a Ru complex and be subsequently released as O2 exclusively as oxidants that damage essential biomolecules,1–3 with concomitant formation of a Ru–H intermediate (Scheme but recent reports have shown that H2O2 can also perform 1A, blue arrows). -

Physical Sciences Content
Foreword In order to improve learning outcomes the Department of Basic Education conducted research to determine the specific areas that learners struggle with in Grade 12 examinations. The research included a trend analysis by subject experts of learner performance over a period of five years as well as learner examination scripts in order to diagnose deficiencies or misconceptions in particular content areas. In addition, expert teachers were interviewed to determine the best practicesto ensure mastery of thetopic by learners and improve outcomes in terms of quality and quantity. The results of the research formed the foundation and guiding principles for the development of the booklets. In each identified subject, key content areas were identified for the development of material that will significantly improve learner's conceptual understanding whilst leading to improved performance in the subject. The booklets are developed as part of a series of booklets, with each bookletfocussing onlyon one specific challenging topic. The selected content is explained in detail and include relevant concepts from Grades 10 - 12 to ensure conceptual understanding. The main purpose of these booklets is to assist learners to master the content starting from a basic conceptual level of understanding to the more advanced level. The content in each booklets is presented in an easy to understand manner including the use of mind maps, summaries and exercises to support understanding and conceptual progression. These booklets should ideally be used as part of a focussed revision or enrichment program by learners after the topics have been taught in class. The booklets encourage learners to take ownership of their own learning and focus on developing and mastery critical content and skills such as reading and higher order thinking skills. -

Heuristics for Searching Chemical Structures a Thesis Presented To
Heuristics for Searching Chemical Structures A Thesis presented to the Faculty of the Graduate School University of Missouri-Columbia In Partial Fulfillment of the Requirements for the Degree Master of Science by Nandini Basu Dr. Toni Kazic, Thesis Supervisor May 2007 The undersigned, appointed by the Dean of the Graduate School, have examined the thesis entitled HEURISTICS FOR SEARCHING CHEMICAL STRUCTURES presented by Nandini Basu A candidate for the degree of Master of Science And hereby certify that in their opinion it is worthy of acceptance. Dr. Toni Kazic Dr. Chi-Ren Shyu Dr. Mary Schaeffer ACKNOWLEDGMENTS I am deeply indebted to my research advisor Dr. Toni Kazic for giving me an opportunity to resume my research in and complete my Computer Science MS program. Without her guidance, support and encouragement this would not have been possible. I would like to thank Dr. Mary Polaco and Dr. Shyu for serving in my committee and helping me in my thesis. I would like to thank the Klotho team for helping me with structures. And also the other team members Amith, Archana, Avanthi, Bill, Deepthi, Gaurav, Raman, Jiahui, Fatten, Meeta and Michael. My friends made a difficult journey lot of fun and much easier, especially Cathy, Charu, Deepak, Karan, Karthik, Paco, Nikunj, Raj, Shiva, Shraddha, Sundeep and Vibha. All my relatives gave me constant encouragement and I would like to take this opportunity to thank them. And above all, I would like to thank my father Prabhat Kumar Basu, my mother Minoti Basu, my brother Pradipta Kumar Basu and my sister-in-law Sarmistha Basu.