Perfluorocarbons in the Global Atmosphere
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

12E4968079.Pdf
Fluorocarbon compatibilized gold-silica nanocomposites for recyclable regioselective hydroamination of alkynes in a fluorous biphasic system Item Type Conference Paper Authors Merican, Zulkifli; Vu, Bao Khanh; Solovyeva, Vera; Rodionov, Valentin; Khe, Cheng Seong; Rajalingam, Sokkalingam; Vasant, Pandian Citation Merican Z, Vu BK, Solovyeva VA, Rodionov VO, Khe CS, et al. (2016) Fluorocarbon compatibilized gold-silica nanocomposites for recyclable regioselective hydroamination of alkynes in a fluorous biphasic system. Available: http:// dx.doi.org/10.1063/1.4968079. Eprint version Publisher's Version/PDF DOI 10.1063/1.4968079 Publisher AIP Publishing Rights This article may be downloaded for personal use only. Any other use requires prior permission of the author and AIP Publishing. The following article appeared in Merican, Z., Vu, B.K., Solovyeva, V.A., Rodionov, V.O., Khe, C.S., Rajalingam, S. and Vasant, P., 2016, November. Fluorocarbon compatibilized gold-silica nanocomposites for recyclable regioselective hydroamination of alkynes in a fluorous biphasic system. In 4TH INTERNATIONAL CONFERENCE ON FUNDAMENTAL AND APPLIED SCIENCES (ICFAS2016) (Vol. 1787, No. 1, p. 030014). AIP Publishing and may be found at http://aip.scitation.org/doi/abs/10.1063/1.4968079. Download date 27/09/2021 23:14:23 Link to Item http://hdl.handle.net/10754/622088 Fluorocarbon compatibilized gold-silica nanocomposites for recyclable regioselective hydroamination of alkynes in a fluorous biphasic system Zulkifli Merican, Bao Khanh Vu, Vera A. Solovyeva, Valentin O. Rodionov, Cheng Seong Khe, Sokkalingam Rajalingam, and Pandian Vasant Citation: 1787, 030014 (2016); doi: 10.1063/1.4968079 View online: http://dx.doi.org/10.1063/1.4968079 View Table of Contents: http://aip.scitation.org/toc/apc/1787/1 Published by the American Institute of Physics Fluorocarbon Compatibilized Gold-Silica Nanocomposites For Recyclable Regioselective Hydroamination of Alkynes In A Fluorous Biphasic System Zulkifli Merican1, a Bao Khanh Vu2, Vera A. -

Hexafluoroethane
Common Name: HEXAFLUOROETHANE CAS Number: 76-16-4 RTK Substance number: 0989 DOT Number: UN 2193 Date: May 1986 Revision: March 1999 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Hexafluoroethane can affect you when breathed in. * If you think you are experiencing any work-related health * Contact can cause severe skin and eye irritation and burns problems, see a doctor trained to recognize occupational with possible eye damage. diseases. Take this Fact Sheet with you. * Breathing Hexafluoroethane can irritate the nose and throat. WORKPLACE EXPOSURE LIMITS * Breathing Hexafluoroethane can irritate the lungs causing No occupational exposure limits have been established for coughing and/or shortness of breath. Higher exposures Hexafluoroethane. This does not mean that this substance is can cause a build-up of fluid in the lungs (pulmonary not harmful. Safe work practices should always be followed. edema), a medical emergency, with severe shortness of breath. * Large amounts of Hexafluoroethane will decrease the * Exposure to the liquified gas can cause frostbite. amount of available Oxygen. Oxygen content should be * Hexafluoroethane can cause headache, nausea, fatigue, routinely tested to ensure that it is at least 19% by volume. mood changes, dizziness, unconsciousness and death. * Very high exposure can cause suffocation from lack of WAYS OF REDUCING EXPOSURE Oxygen. It may also cause the heart to beat irregularly * Where possible, enclose operations and use local exhaust (arrhythmia) or to stop, which can be fatal. ventilation at the site of chemical release. If local exhaust ventilation or enclosure is not used, respirators should be IDENTIFICATION worn. Hexafluoroethane is a colorless and odorless gas, or liquid * Wear protective work clothing. -

Low-Surface-Energy Fluoromethacrylate Block Copolymers with Patternable Elements
Chem. Mater. 2000, 12, 33-40 33 Low-Surface-Energy Fluoromethacrylate Block Copolymers with Patternable Elements Shu Yang, Jianguo Wang,† Kenji Ogino,‡ Suresh Valiyaveettil,§ and Christopher K. Ober* Department of Materials Science and Engineering, Cornell University, Bard Hall, Ithaca, New York 14853-1501 Received April 26, 1999. Revised Manuscript Received October 19, 1999 A series of block polymers were synthesized from tetrahydropyranyl methacrylate (THPMA) and several fluorinated methacrylates. By changing both the length of the fluorinated side chain and the nature of its end group, the surface properties of these polymers were greatly influenced. The polymers with perfluoroheptyl groups showed the lowest surface tension, ∼7 mN/m, whereas those with perfluoropropyl groups showed surface tension around 12 mN/m. The surface tension changed dramatically when the perfluorinated side chain with a CF2H end group was capped, increasing to 18 mN/m for three -CF2- units. The relative volume ratios of different block copolymers did not affect the resulting surface energy. After thermal decomposition of labile THP- groups in these polymers, acid and acid anhydride groups were produced. Thermal cross-linking of these groups increased mechanical robustness and led to better surface stability and adhesion of the polymer to the substrate. Because these polymers were designed for their lithographic abilities, some aspects of their photoimaging properties are discussed. Introduction surfactants, or emulsifiers,2 some of which are soluble in supercritical carbon dioxide.3,4 A monolayer of these Precise control of the surface properties of polymeric materials, properly organized, will make a very low- materials by means of self-organization is a major energy surface. -

JTEFT-04-00146.Pdf
Journal of Textile Engineering & Fashion Technology Research Article Open Access Process intensification of fluorocarbon-free and fluorocarbon-based water repellent finishes on cotton knit fabrics Abstract Volume 4 Issue 3 - 2018 The capabilities of the fluorocarbon-free alkyl urethane based resin, was analyzed Kawser Parveen Chowdhury on cotton fabric. In this study, both single jersey and double jersey knit structured Department of Wet Process Engineering, Bangladesh University fabrics were taken to evaluate the performance of different water repellent finishes of Textiles, Bangladesh; on fabrics properties. The performance of the fluorocarbon-free alkyl urethane based resin and fluorocarbon based water repellent chemicals were evaluated and compared Correspondence: Kawser Parveen Chowdhury, Assistant at different formulations. The effectiveness of water repellency of the finished fabrics Professor, Department of Wet Process Engineering, Bangladesh were evaluated by AATCC 127 hydrostatic head test method and by ISO 4920:2012 University of Textiles, Address: 92, Shaheed Tajuddin Ahmed spray rating test method. To assess the performance of water repellent finished knit Avenue, Tejgaon, Dhaka-1208, Bangladesh, Tel +8801716167777, fabrics, GSM, bursting strength test, stiffness, color fastness to wash, color fastness to Email [email protected] sea water, color fastness to saliva, color fastness to rubbing, color fastness to light were done according to ISO and ASTM method. The results showed that the fluorocarbon- Received: May 25, 2018 | Published: June 06, 2018 free alkyl urethane based resin treated fabrics exhibited competitive result on water repellency, other physical and chemicals properties. The water repellent finish type and concentration were very important criteria to obtain good water repellency. -

Finding Refrigerant Leaks with the Chempro100i
Application Note: 108 Finding Refrigerant Leaks with the ChemPro100i The ChemPro100i is a great sniffer for halocarbon refrigerants, often generically referred to as “Freon” because of its multiple sensors and broad sniffing capability. As refrigerant sniffers are only carried by a small subset of first responders, the ChemPro100i can fill the role for those that don’t have a refrigerant sniffer. As a bonus, if someone suspects that it may be a refrigerant leak, and it turns out to be something else, the wide range of detectable gases and vapors seen by the ChemPro100i means that it will most likely find the unexpected gas/vapor too. The ChemPro100i’s Sensors The ChemPro100i uses a suite of seven sensors including an aspirated Ion Mobility Spectroscopy (IMS), five metal oxide sensors and a field effect sensor to detect, characterize, and even identify, some gases and vapors. Using this suite of sensors, the ChemPro100i can find halocarbon refrigerant leaks using its “Trend” or “sniffer” screen. As one gets closer to the refrigerant leak, the trend line will Using other Sniffers for increase. This is a non-quantifiable reading Refrigerants that does not directly correlate with parts per Most dedicated refrigerant detectors use a million (ppm), but the fast response of the Metal Oxide Sensor (MOS) that is doped to ChemPro100i to refrigerants provides one with be relatively specific to the halogenated the means of quickly finding a refrigerant leak. hydrocarbons (halogens) that are the hallmark of most refrigerants. MOS sensors Increase in the Trend line leads you to the leak are non-linear and not suitable for quantification of refrigerants, but they provide sensitive and fast response to halocarbon refrigerants. -

Hfcs for THERMAL INSULATION
HFCs FOR THERMAL INSULATION Visit our web site www.fluorocarbons.org A SOLUTION ADDRESSING THE CLIMATE CHANGE CHALLENGE EFCTC - European FluoroCarbon Technical Committee Avenue E.van Nieuwenhuyse 4 B-1160 Brussels - Phone : +32 2 676 72 11 HFCs FOR THERMAL INSULATION Visit our web site www.fluorocarbons.org A SOLUTION ADDRESSING THE CLIMATE CHANGE CHALLENGE EFCTC - European FluoroCarbon Technical Committee Avenue E.van Nieuwenhuyse 4 B-1160 Brussels - Phone : +32 2 676 72 11 Progress for our quality of life… Could we do without heating or air conditioning? …and climate protection Refrigeration, air conditioning and heating are often essential to life particularly in public areas such as The greenhouse effect to a great extent determines the climate in hospitals and laboratories, for food products on earth. Growth in emissions of greenhouse gases associated with in the cold chain, for medical and computer equipment. human activities threa tens the climate balance. Carbon dioxide (CO2) - the main greenhouse gas - is emitted when fossil fuels are burnt to But we have also to consider the impact of produce energy and increasing energy demands have led to rapid growth the wide-spread use of these services on in the amount of CO2 in the atmosphere. Heating, air conditioning the global environment. and refrigeration have contributed to this growth. Roof If no action is taken at all, greenhouse gas(*) emissions could be expected to further increase in EU Member States by 17% 22% between 1990 and 2010, while the target set by the Kyoto Protocol for the period is to reduce the emissions by 8%. -

Halocarbon 32 (Difluoromethane) CH 2F2
Halocarbon 32 (Difluoromethane) CH 2F2 Grade ULSI 4N ULTIMA 4N8 Purity, % 99.99 99.998 Nitrogen ≤40 ppmv ≤5 ppmv Oxygen ≤10 ppmv ≤2 ppmv Carbon Dioxide ≤15 ppmv ≤1 ppmv Methane ≤2 ppmv ≤1 ppmv Water ≤5 ppmv ≤1 ppmv Carbon Tetrafluoride ≤5 ppmv ≤1 ppmv Other Organics ≤100 ppmv ≤10 ppmv Carbon Monoxide ≤1 ppmv Acidity as HF ≤0.1 ppmw • A lot analysis is provided for each order – Individual analysis is also available upon request. • Other Organics = H 12 , H 22 , H 23 , H 134a Internal Volume Liters 43.8 17.1 7.3 R E Cylinder Sizes >> QF GF UF D N I L lbs 64 25 11 Y Content C kg 29.1 11.35 5 Change Point* lbs 4.3 1.7 0.7 *Recommended Cylinder Change Point at NTP, based on Phase Break, or the amount of product left in the cylinder when the liquid phase has completely evaporated and only gaseous product is left (estimate based on ideal gas behavior). DOT Shipping Name Difluoromethane Shipped as P I DOT Classification 2.1 (Flammable Gas) H S Liquefied DOT Label FLAMMABLE GAS Gas UN Number UN 3252 Cylinder Pressure 207 psig Temp, °C 0.0 15.5 21.0 32.2 43.3 A Vapor T A @NTP 15.6 atm Press, psig 102 172 207 279 372 D 3 Pressure L Specific Volume 0.45 m /kg Temp, °F 32 60 70 90 110 A 3 C NTP = 21°C or 70°F and 101.3 kPa or 1 atm I @NTP 7.2 ft /lb N H CAS No 75-10-5 C E T CGA/DISS/JIS 350/724/W22-14L Molecular Weight 52 g/mol Nominal Diameter (OD)xHeight* Material of Construction Cylinder Treatment cm Inches Cylinder Valve ® QF ULTRA-LINE 23x130/134/143 9x51/52.5/56 CS SS ® GF ULTRA-LINE 23x66/70/79 9x26/27.5/31 CS SS ® UF ULTRA-LINE 15x51/55/64 6x19/20.5/24 CS SS *Height is reported as the distance from the bottom of the cylinder to the cylinder neck/ center of the valve outlet/ top of the handwheel CS: Carbon Steel SS: Stainless Steel WARNING: This product can expose you to chemicals including Carbon Monoxide, which is known to the State of California to cause birth defects or other reproductive harm. -

Safety Data Sheet Tetrafluoromethane
Safety Data Sheet Tetrafluoromethane Section 1: Product and Company Identification Middlesex Gases & Technologies 292 Second Street P.O. Box 490249 Everett, MA 02149 (617) 387-5050 (800) 649-6704 Fax (617) 387-3537 http://www.middlesexgases.com/ Product Code: Tetrafluoromethane Section 2: Hazards Identification Warning Hazard Classification: Gases Under Pressure Hazard Statements: Contains gas under pressure; may explode if heated Precautionary Statements Storage: Protect from sunlight. Store in well-ventilated place. Section 3: Composition/Information on Ingredients CAS # 75-73-0 Middlesex Gases & Technologies page 1 of 5 Generated by the SDS Manager from AsteRisk, LLC. All Rights Reserved Generated: 06/01/2015 Chemical Substance Chemical Trade Names Family TETRAFLUOROMETHANE halogenated, CARBON TETRAFLUORIDE; CARBON FLUORIDE (CF4); CARBON FLUORIDE; FC 14; aliphatic PERFLUOROMETHANE; R 14; R 14 (REFRIGERANT); METHANE, TETRAFLUORO-; FREON 14; TETRAFLUOROCARBON; UN 1982; CF4 Section 4: First Aid Measures Skin Contact Eye Contact Ingestion Inhalation Note to Physicians If frostbite or freezing occur, Wash eyes immediately with If a large If adverse effects occur, remove to For immediately flush with plenty of large amounts of water, amount is uncontaminated area. Give artificial inhalation, lukewarm water (105-115 F; 41-46 occasionally lifting upper and swallowed, get respiration if not breathing. If consider C). DO NOT USE HOT WATER. If lower lids, until no evidence of medical breathing is difficult, oxygen should oxygen. warm water is not available, gently chemical remains. Get attention. be administered by qualified wrap affected parts in blankets. Get medical attention immediately. personnel. Get immediate medical immediate medical attention. attention. Section 5: Fire Fighting Measures Suitable Extinguishing Media Products of Combustion Protection of Firefighters Non-flammable. -

Factsheet Fluorocarbons (Pfcs)
Factsheet Fluorocarbons (PFCs) Sympatex Technologies GmbH PTFE-free Sympatex membrane The Sympatex membrane is PTFE-free which means that it does not contain any fluorine compounds. It is made of perfectly safe polyether/ester, a compound of polyester and polyether molecules which makes it absolutely environmentally friendly and kind on the skin and also means it can be recycled like a PET bottle. Other membranes such as Gore-Tex and eVent are made of PTFE (polytetrafluoroethylene) whose manufacture, disposal, heating and incineration may release PFCs (per and polyfluorinated chemicals / fluorine compounds) which are regarded as ecologically problematic. The performance of fluorine-free polyester membranes and PTFE-based membranes are very similar in terms of them being windproof, breathable and waterproof. Water-repellent impregnations / General facts To meet requirements relating to functionality and care, the outer materials used in outdoor clothing generally have a water-repellent treatment (impregnation) on the outside and a membrane on the inside. The Sympatex membrane ensures that functional clothing is waterproof. Water-repellent treatments also ensure that the textile has durable water-repellent properties. A DWR (durable water repellent) treatments is an extremely thin coating of the individual fibres which ensures that water simply forms beads and falls off. This treatment prevents or delays the ingress of water into the outer material. The result of this is that the garment does not absorb water, it does not gain weight, the -
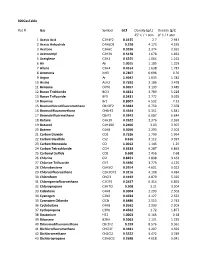
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

And Perfluoroalkyl Substances (PFAS): Sources, Pathways and Environmental Data
Poly- and perfluoroalkyl substances (PFAS): sources, pathways and environmental data Chief Scientist’s Group report August 2021 We are the Environment Agency. We protect and improve the environment. We help people and wildlife adapt to climate change and reduce its impacts, including flooding, drought, sea level rise and coastal erosion. We improve the quality of our water, land and air by tackling pollution. We work with businesses to help them comply with environmental regulations. A healthy and diverse environment enhances people's lives and contributes to economic growth. We can’t do this alone. We work as part of the Defra group (Department for Environment, Food & Rural Affairs), with the rest of government, local councils, businesses, civil society groups and local communities to create a better place for people and wildlife. Published by: Author: Emma Pemberton Environment Agency Horizon House, Deanery Road, Environment Agency’s Project Manager: Bristol BS1 5AH Mark Sinton www.gov.uk/environment-agency Citation: Environment Agency (2021) Poly- and © Environment Agency 2021 perfluoroalkyl substances (PFAS): sources, pathways and environmental All rights reserved. This document may data. Environment Agency, Bri be reproduced with prior permission of the Environment Agency. Further copies of this report are available from our publications catalogue: www.gov.uk/government/publications or our National Customer Contact Centre: 03708 506 506 Email: research@environment- agency.gov.uk 2 of 110 Research at the Environment Agency Scientific research and analysis underpins everything the Environment Agency does. It helps us to understand and manage the environment effectively. Our own experts work with leading scientific organisations, universities and other parts of the Defra group to bring the best knowledge to bear on the environmental problems that we face now and in the future.