Cyanide and Central Nervous System a Study with Focus on Brain Dopamine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acute Toxicity of Cyanide in Aerobic Respiration: Theoretical and Experimental Support for Murburn Explanation
BioMol Concepts 2020; 11: 32–56 Research Article Open Access Kelath Murali Manoj*, Surjith Ramasamy, Abhinav Parashar, Daniel Andrew Gideon, Vidhu Soman, Vivian David Jacob, Kannan Pakshirajan Acute toxicity of cyanide in aerobic respiration: Theoretical and experimental support for murburn explanation https://doi.org/10.1515/bmc-2020-0004 received January 14, 2020; accepted February 19, 2020. of diffusible radicals and proton-equilibriums, explaining analogous outcomes in mOxPhos chemistry. Further, we Abstract: The inefficiency of cyanide/HCN (CN) binding demonstrate that superoxide (diffusible reactive oxygen with heme proteins (under physiological regimes) is species, DROS) enables in vitro ATP synthesis from demonstrated with an assessment of thermodynamics, ADP+phosphate, and show that this reaction is inhibited kinetics, and inhibition constants. The acute onset of by CN. Therefore, practically instantaneous CN ion-radical toxicity and CN’s mg/Kg LD50 (μM lethal concentration) interactions with DROS in matrix catalytically disrupt suggests that the classical hemeFe binding-based mOxPhos, explaining the acute lethal effect of CN. inhibition rationale is untenable to account for the toxicity of CN. In vitro mechanistic probing of Keywords: cyanide-poisoning; murburn concept; CN-mediated inhibition of hemeFe reductionist systems aerobic respiration; ATP-synthesis; hemoglobin; was explored as a murburn model for mitochondrial cytochrome oxidase; mitochondria; diffusible reactive oxidative phosphorylation (mOxPhos). The effect of CN oxygen species (DROS). in haloperoxidase catalyzed chlorine moiety transfer to small organics was considered as an analogous probe for phosphate group transfer in mOxPhos. Similarly, Introduction inclusion of CN in peroxidase-catalase mediated one- electron oxidation of small organics was used to explore Since the discovery of the dye Prussian blue (ferric electron transfer outcomes in mOxPhos, leading to water ferrocyanide, Fe7[CN]18) and the volatile prussic acid formation. -

Hazardous Chemicals in Secondhand Marijuana Smoke
Hazardous Chemicals in Secondhand Marijuana Smoke “The following 33 marijuana smoke constituents included in Table 1 are listed under 33 Chemicals Proposition 65 as causing cancer: acetaldehyde, acetamide, acrylonitrile, 4- aminobiphenyl, arsenic, benz[a]anthracene, benzene, benzo[a]pyrene, That Can benzo[b]fluoranthene, benzo[j]fluoranthene, benzo[k]fluoranthene, benzofuran, 1,3- butadiene, cadmium, carbazole, catechol, chromium (hexavalent compounds), Cancer chrysene, dibenz[a,h]anthracene, dibenz[a,i]pyrene, dibenzo[a,e]pyrene, “Many of the chemical diethylnitrosamine, dimethylnitrosamine, formaldehyde, indeno[1,2,3,-c,d]pyrene, constituents that have been isoprene, lead, mercury, 5-methylchrysene, naphthalene, nickel, pyridine, and identified in marijuana smoke quinoline.” are carcinogens.” 2009 OEHHA document, Evidence on the Carcinogenicity of Marijuana Smoke Hydrogen Cyanide interferes with the normal use of oxygen by nearly every organ of Hydrogen the body. Exposure to hydrogen cyanide (AC) can be rapidly fatal. It has whole-body (systemic) effects, particularly affecting those organ systems most sensitive to low Cyanide oxygen levels: the central nervous system (brain), the cardiovascular system (heart Is the same chemical used for and blood vessels), and the pulmonary system (lungs). Hydrogen cyanide (AC) is a chemical weapons. chemical warfare agent (military designation, AC). Ammonia gas is a severe respiratory tract irritant. Can cause severe irritation of the Ammonia nose and throat. Can cause life-threatening accumulation of fluid in the lungs Household cleaner used on (pulmonary edema). Symptoms may include coughing, shortness of breath, difficult floors and toilets. There is 3 breathing and tightness in the chest. Symptoms may develop hours after exposure times more in secondhand and are made worse by physical effort. -
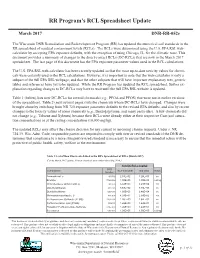
RR Program's RCL Spreadsheet Update
RR Program’s RCL Spreadsheet Update March 2017 RR Program RCL Spreadsheet Update DNR-RR-052e The Wisconsin DNR Remediation and Redevelopment Program (RR) has updated the numerical soil standards in the August 2015 DNR-RR- 052b RR spreadsheet of residual contaminant levels (RCLs). The RCLs were determined using the U.S. EPA RSL web- calculator by accepting EPA exposure defaults, with the exception of using Chicago, IL, for the climatic zone. This documentThe U.S. provides EPA updateda summary its Regionalof changes Screening to the direct-contact Level (RSL) RCLs website (DC-RCLs) in June that2015. are To now reflect in the that March 2017 spreadsheet.update, the The Wisconsin last page ofDNR this updated document the has numerical the EPA exposuresoil standards, parameter or residual values usedcontaminant in the RCL levels calculations. (RCLs), in the Remediation and Redevelopment program’s spreadsheet of RCLs. This document The providesU.S. EPA a RSL summary web-calculator of the updates has been incorporated recently updated in the Julyso that 2015 the spreadsheet.most up-to-date There toxicity were values no changes for chemi - cals madewere certainlyto the groundwater used in the RCLs,RCL calculations. but there are However, many changes it is important in the industrial to note that and the non-industrial web-calculator direct is only a subpartcontact of the (DC) full RCLsEPA RSL worksheets. webpage, Tables and that 1 andthe other 2 of thissubparts document that will summarize have important the DC-RCL explanatory changes text, generic tablesfrom and the references previous have spreadsheet yet to be (Januaryupdated. -

Qualitative Chemical Measures to Indirectly Assess Quality of Natural/Uncured, Organic Bacon Compared to Traditionally Cured Bacon
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2009 Qualitative chemical measures to indirectly assess quality of natural/uncured, organic bacon compared to traditionally cured bacon. Charlwit Kulchaiyawat Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Animal Sciences Commons Recommended Citation Kulchaiyawat, Charlwit, "Qualitative chemical measures to indirectly assess quality of natural/uncured, organic bacon compared to traditionally cured bacon." (2009). Graduate Theses and Dissertations. 10999. https://lib.dr.iastate.edu/etd/10999 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Qualitative chemical measures to indirectly assess quality of natural/uncured, organic bacon compared to traditionally cured bacon by Charlwit Kulchaiyawat A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Meat Science Program of Study Committee: Joseph Sebranek, Major Professor James Dickson Mark Honeyman Lester Wilson Iowa State University Ames, Iowa 2009 Copyright © Charlwit Kulchaiyawat 2009. All rights reserved. ii TABLE OF CONTENTS LIST OF TABLES iii ABSTRACT -

Nitric Oxide.Vp
CHAPTER 5 Maple Leaf Foods, Toronto, Canada Brown Foundation Institute of Molecular Medicine, The University of Texas Health Science Center, Houston, TX 77030 ESPITE the many published reports on the beneficial properties of Dnitrite and nitrate in physiology, nitrite and nitrate in cured and pro- cessed meats continues to be perceived as harmful. The previous chapter revealed that certain foods, particularly green leafy vegetables are natu- rally enriched in nitrite and nitrate from growing in soil. However, en- riching meats with nitrite or nitrate during curing is perceived as harmful and advocated by some groups to be eliminated completely. The use of pure sodium nitrate in curing is now only a minor practice in the United States. The ingoing levels of sodium nitrite have been tightly controlled by the Food Safety and Inspection Service (FSIS) of the U.S. Depart- ment of Agriculture. To satisfy consumer demands for no added nitrite processed meats, efforts have recently been taken to creatively adjust the meat curing process by employing “nitrite free” organic vegetable pow- ders instead of directly adding sodium nitrite salts. Although the end re- sult is production of nitrite from the nitrate contained in the vegetable powders, a more “natural” or “organic” approach seems to appeal to con- sumers. This is primarily due to the public perception of nitrite and ni- trate. Reports about methemoglobinemia in infants (blue baby syndrome) caused by drinks or food prepared with nitrate-rich (and bac- terially contaminated) well water and vegetables, intentional and occu- pational intoxications in adults, increasing nitrate levels in soil and lakes as a result of fertilizer overuse, and the formation of potentially carcino- genic N-nitrosamines all contribute to the negative image that nitrite and nitrate have held in recent years. -

The Use of Sodium Cyanide in Wildlife Damage Management
Human Health and Ecological Risk Assessment for the Use of Wildlife Damage Management Methods by USDA-APHIS-Wildlife Services Chapter VII THE USE OF SODIUM CYANIDE IN WILDLIFE DAMAGE MANAGEMENT May 2017 Peer Reviewed Final October 2019 THE USE OF SODIUM CYANIDE IN WILDLIFE DAMAGE MANAGEMENT EXECUTIVE SUMMARY USDA-APHIS Wildlife Services (WS) uses sodium cyanide (NaCN) to manage coyotes, red foxes, gray foxes, arctic foxes, and wild dogs that prey upon livestock, poultry, and federally designated threatened and endangered species or animals that are vectors of disease. This human health and ecological risk assessment is an evaluation of the risks to human health, nontarget animals, and the environment from NaCN use by WS. WS uses the M-44, the name for the ejector device that delivers a single dose NaCN from a capsule, to target canids. The M-44 is spring-activated and is actuated when an animal pulls up on the capsule holder; a plunger propelled by the spring breaks through a capsule with dry NaCN to deliver the contents into the mouth of an animal. The WS applicator baits the M-44 capsule holder sides to attract target canids. Sodium cyanide reacts rapidly with moisture in the mouth or mucus membranes of the nose and eyes to form hydrogen cyanide (HCN), a toxicant. One NaCN capsule contains enough cyanide to be lethal to animals through oral contact, inhalation contact, and moist dermal pathway contact. WS annually averaged the known take of 13,959 target canids and 362 nontarget species with NaCN between FY11 and FY15, recording 1,548,000 Method Nights with M-44s in 17 States. -

Cyanide CAS# 74-90-8, 143-33-9, 151-50-8, 592-01-8, 544-92-3, 506-61-6, 460-19-5, 506-77-4
Cyanide CAS# 74-90-8, 143-33-9, 151-50-8, 592-01-8, 544-92-3, 506-61-6, 460-19-5, 506-77-4 Division of Toxicology ToxFAQsTM September 2004 This fact sheet answers the most frequently asked health questions (FAQs) about cyanide. For more information, call the ATSDR Information Center at 1-888-422-8737. This fact sheet is one in a series of summaries about hazardous substances and their health effects. It is important you understand this information because this substance may harm you. The effects of exposure to any hazardous substance depend on the dose, the duration, how you are exposed, personal traits and habits, and whether other chemicals are present. HIGHLIGHTS: Exposure to high levels of cyanide harms the brain and heart, and may cause coma and death. Exposure to lower levels may result in breathing difficulties, heart pains, vomiting, blood changes, headaches, and enlargement of the thyroid gland. Cyanide has been found in at least 471 of the 1,647 National Priorities List sites identified by the Environmental Protection Agency (EPA). What is cyanide? ‘ Most cyanide in surface water will form hydrogen Cyanide is usually found joined with other chemicals to form cyanide and evaporate. compounds. Examples of simple cyanide compounds are ‘ Cyanide in water does not build up in the bodies of fish. hydrogen cyanide, sodium cyanide and potassium cyanide. ‘ Cyanides are fairly mobile in soil. Once in soil, cyanide Certain bacteria, fungi, and algae can produce cyanide, and can be removed through several processes. Some cyanide cyanide is found in a number of foods and plants. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Hydrogen Cyanide Fact Sheet What Is Hydrogen Cyanide? at Room Temperature, Hydrogen Cyanide Is a Volatile, Colorless-To-Blue Liquid (Also Called Hydrocyanic Acid)
Hydrogen Cyanide Fact Sheet What is hydrogen cyanide? At room temperature, hydrogen cyanide is a volatile, colorless-to-blue liquid (also called hydrocyanic acid). It rapidly becomes a gas that can produce death in minutes if breathed. Hydrogen cyanide is used in making fibers, plastics, dyes, pesticides, and other chemicals, and as a fumigant to kill rats. It is also used in electroplating metals and in developing photographic film. What immediate health effects can be caused by exposure to hydrogen cyanide? Breathing small amounts of hydrogen cyanide may cause headache, dizziness, weakness, nausea, and vomiting. Larger amounts may cause gasping, irregular heartbeats, seizures, fainting, and even rapid death. Generally, the more serious the exposure, the more severe the symptoms. Similar symptoms may be produced when solutions of hydrogen cyanide are ingested or come in contact with the skin. Can hydrogen cyanide poisoning be treated? The treatment for cyanide poisoning includes breathing pure oxygen, and in the case of serious symptoms, treatment with specific cyanide antidotes. Persons with serious symptoms will need to be hospitalized. Call your doctor or the Emergency Department if you develop any unusual signs or symptoms within the next 24 hours, especially: difficulty breathing, shortness of breath, or chest pain confusion or fainting increased pain or a discharge from your eyes increased redness, pain, or a pus-like discharge in the area of a skin burn Are any future health effects likely to occur? A single small exposure from which a person recovers quickly is not likely to cause delayed or long term effects. After a serious exposure, a patient may have brain or heart damage. -

1997-11-12 Cyanide Compounds As Federal Hazardous Air Pollutant
CYANIDE COMPOUNDS Cyanide compounds are federal hazardous air pollutant and were identified as toxic air contaminants in April 1993 under AB 2728. CAS Registry Numbers: cyanide 57-12-5 CN hydrocyanic acid 74-90-8 HCN Molecular Formulas: CN- HCN The cyanide ion (CN-) combines with other chemicals and exhibits many characteristics that are similar to those of the halogen and azide ions. The molecular weight of cyanide is 26.02. It has an unusually strong tendency to form complex ions with metal ions which may be easily oxidized and thermally unstable. Cyanides are flammable by chemical reaction with heat, moisture, and acid. Many cyanides easily release the very toxic hydrocyanic acid (HCN) which has the odor of bitter almonds. Hydrocyanic acid (hydrogen cyanide) is a liquid at temperatures below 26.5 oC, and its boiling point is so close to room temperature that evaporation of liquid HCN evolves to vapor. Liquid or gaseous HCN is extremely soluble in water. Hydrocyanic acid is miscible with alcohol and slightly soluble in ether. Many organic nitriles can be very reactive under the right conditions and when heated to decomposition or on contact with acid, acid fumes, water or steam, toxic and flammable vapors of CN- are emitted (HSDB, 1991). Physical Properties of Hydrocyanic Acid Synonyms for cyanide: carbon nitride ion (CN1-); cyanide anion; isocyanide Synonyms for hydrocyanic acid: hydrogen cyanide; anammonide; formonitrile; cyclon; prussic acid Hydrocyanic acid Molecular Weight 27.03 Boiling Point: 25.6 oC Melting Point: -13.4 oC Density (liquid): 0.699 at 22 oC Density (gas): 0.901 at 22 oC Vapor Density: 0.94 (air = 1) Vapor Pressure: 630 mm Hg at 20 oC Heat of Fusion: 74.38 Log Octanol/Water Partition Coefficient: 1.07 Conversion Factor (HCN): 1 ppm = 1.1 mg/m3 (HSDB, 1991; Merck, 1989; Sax, 1989; U.S. -

Hydrogen Cyanide and Cyanides: Human Health Aspects
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 61 HYDROGEN CYANIDE AND CYANIDES: HUMAN HEALTH ASPECTS Please note that the layout and pagination of this pdf file are not identical to the version in press First draft prepared by Prof. Fina Petrova Simeonova, Consultant, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria; and Dr Lawrence Fishbein, Fairfax, Virginia, USA Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2004 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management -

United States Patent Office Patented Sept
3,149,57 United States Patent Office Patented Sept. 15, 1964 2 Gold alloy plates may also be obtained from this bath 3,149,957 ACD GOLD PATNG by incorporating into the bath certain metal salts, com Edward A. Parker, Cransion, and James A. Powers, East plexes, or chelates, because such other metals plate out Providence, R.I., assignors to Technie, Inc., Providence, with the gold. Among such compounds may be men R.E., a corporation of Rhode island tioned antimony tartrate, nickel or cadmium ethylene No Drawing. Filed Apr. 27, 1959, Ser. No. 808,913 diamine tetra-acetic acid. Similar compounds of indium, 1. Claims. (C. 204-46) zinc, copper, iron may be used to obtain alloy plates. It is, therefore, a primary object of the present inven The present invention relates to the electrolytic de tion to provide an aqueous acid solution from which gold position of gold on surfaces and more particularly to the O may be electrodeposited in which the harmful conse electrodeposition of gold and gold alloys from solutions quences of the formation of free hydrogen cyanide are of the same onto objects to develop a gold or gold alloy eliminated. appearance. It is another object of this invention to provide a bath It has been customary for many years in the electro for the deposition of substantially 24 karat gold electro plating of gold and gold alloys to use a bath containing plate which is characterized by higher hardness, greater an alkali metal aurocyanide, with or without other soluble wear resistance, and increased speed of plating than that metal salts.