Abstract Book
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Universities Receiving the Grant by Region Japan Hokkaido
List of Universities receiving the Grant by Region Japan Hokkaido University Hokkaido Kokushikan University Tokyo Tohoku Fukushi University Miyagi Musashino University Tokyo Tohoku University Miyagi Soka University Tokyo Akita International University Akita Showa Womens University Tokyo Yamagata University Yamagata J. F. Oberlin University Tokyo Fukushima University Fukushima Toho Gakuen College of Drama and Music Tokyo University of Tsukuba Ibaraki National Defense Academy of Japan Kanagawa Heisei International University Saitama Ferris University Kanagawa Meikai University Chiba Bunkyo University Kanagawa Tokyo University of Science Chiba Kanto Gakuin University Kanagawa Ryutsu Keizai University Chiba Keio University Kanagawa Nihon University College of Art Tokyo University of Niigata Prefecture Niigata Kaetsu University Tokyo University of Toyama Toyama University of Tokyo College of Arts and Science Tokyo Takaoka University of Law Toyama University of Tokyo Graduate School of Interdisciplinary Studies Tokyo Hokuriku University Ishikawa Waseda Unviersity Graduate School Tokyo Yamanashi Gakuin University Yamanashi Tokyo Gakugei University Tokyo Nihon University College of International Relations Shizuoka Hitotsubashi University Tokyo Aichi University Aichi Tokyo University of Foreign Studies Tokyo Aichi Prefectural University Aichi Meiji University Tokyo Ritsumeikan University Kyoto Teikyo University Tokyo Ryukoku University Kyoto Aoyama Gakuin University Tokyo Heian Jogakuin University Kyoto Nihon University College of Humanities and -

The Birth of Fullerene Chemistry: Harold W. Kroto Discusses New Lines of Buckyball Research in a Science Watchm Interview
Current Comments@ EUGENE GARFIELD INSTITUTE FOB SCIENTIFIC !NFORMATION@ I 3501 MARKET ST, PHILADELPHIA, PA I W04 The Birth of Fullerene Chemistry: Harold W. Kroto Discusses New Lines of Buckyball Research in a Science Watchm Interview Number 37 September 13, 1993 A Star Is Born: Discovering the Third Not surprisingly, buckyballs and the new Form of Carbon field of fullerene chemistry have attracted Last week in the engineering and phys- much attention in the press, For example, ics/chemi stry editions of Currenr Contents@ Science selected the buckyball as its Mol- (C@), we published a Citufion Classic@ ecule of the Year in 1991,6 and the Econo- commentary by Harold W. Kroto, Univer- misf called it (be Renaissance Molecule in sity of Sussex, Brighton, EngIand, on the 1992,7 It was first featured in CC in a 1988 1985 Nature paper describing the discov- essay on the most-cited 1985 chemistry pa- ery of buckminsterfullerene, IZ Working pers.~ with a team of colleagues at Rice Univer- h addition, Kroto was interviewed in sity, Houston, led by Richard E, Smalley, Science WaKh@, ISI@’s newsletter ihat Kroto was interested in learning more about &acks quantitative trends in researches The the interstellar formation of long carbon 1992 interview, reprinted below, focused chains in red giant stars. An unexpected on new directions in fullerene research and result of their effort was the serendipitous its applications in various fields. It is a discovery of a third natural form of car- useful companion piece to Kroto’s Cita- bon—the stable Cm molecule named after tion Classic commentary, ] because both R. -

CONICYT Ranking Por Disciplina > Sub-Área OECD (Académicas) Comisión Nacional De Investigación 1
CONICYT Ranking por Disciplina > Sub-área OECD (Académicas) Comisión Nacional de Investigación 1. Ciencias Naturales > 1.6 Ciencias Biológicas Científica y Tecnológica PAÍS INSTITUCIÓN RANKING PUNTAJE USA Harvard University 1 5,000 USA Massachusetts Institute of Technology (MIT) 2 5,000 UNITED KINGDOM University of Oxford 3 5,000 USA Stanford University 4 5,000 UNITED KINGDOM University of Cambridge 5 5,000 USA Johns Hopkins University 6 5,000 USA University of California San Francisco 7 5,000 USA University of Washington Seattle 8 5,000 UNITED KINGDOM University College London 9 5,000 USA Cornell University 10 5,000 CANADA University of Toronto 11 5,000 USA University of Pennsylvania 12 5,000 USA University of California San Diego 13 5,000 DENMARK University of Copenhagen 14 5,000 USA University of Michigan 15 5,000 USA University of California Berkeley 16 5,000 USA University of California Los Angeles 17 5,000 USA Duke University 18 5,000 USA University of California Davis 19 5,000 UNITED KINGDOM Imperial College London 20 5,000 USA Columbia University 21 5,000 USA Yale University 22 5,000 USA University of Minnesota Twin Cities 23 5,000 FRANCE Universite Paris Saclay (ComUE) 24 5,000 USA University of North Carolina Chapel Hill 25 5,000 AUSTRALIA University of Queensland 26 5,000 AUSTRALIA University of Melbourne 27 5,000 USA Washington University (WUSTL) 28 5,000 NETHERLANDS Utrecht University 29 5,000 USA University of Wisconsin Madison 30 5,000 FRANCE Sorbonne Universite 31 5,000 SWEDEN Karolinska Institutet 32 5,000 USA University -

The Science Studio with Sir Harold Kroto
The Science Studio With Sir Harold Kroto ROGER BINGHAM: My guest today on The Science Studio is Sir Harold Kroto. He’s currently at Florida State University, but in 1996 he shared the Nobel Prize in chemistry for the discovery of a new form of carbon, the C60 molecule called Buckminsterfullerene. He’s received the Royal Society’s prestigious Michael Faraday Award, given annually to the scientist who’s done the most to further public communication of science, engineering, or technology in the United Kingdom, although he now lives here. He’s chairman of the board of the Vega Science Trust, which is an educational charity that produces science programs for television. Harry, welcome. SIR HAROLD KROTO: My pleasure. BINGHAM: So give me some sense of where you came from and what’s your family background, how you got into science. KROTO: Well, my parents were refugees from Berlin, and my father got out of Germany the day before the police came to arrest him. My mother wasn’t Jewish but he was, and we ended up in a town called Bolton, which was the center of the industrial revolution as much as anywhere else. Samuel Crompton, the Spinning Jenny and things, the center of the spinning industry, and weaving as well. And I saw that decay as I was a child. I had a great childhood in the sense that I was very happy and went to a good school. My name originally was Krotoschine, and that’s where I - BINGHAM: [Interposing] Where does that come from? KROTO: Well, it’s the German form of - and there’s a Polish form, Krotoschinsky - and it comes from Krotoschin, a town which is now in Poland but was then in east Prussia. -

4.05 Otras Ciencias Agrícolas
CONICYT Ranking por Disciplina > Sub-área OECD (Académicas) Comisión Nacional de Investigación 4. Ciencias Agrícolas > 4.5 Otras Ciencias Agrícolas Científica y Tecnológica PAÍS INSTITUCIÓN RANKING PUNTAJE NETHERLANDS Wageningen University & Research 1 5,000 CHINA Jiangnan University 2 5,000 CHINA South China University of Technology 3 5,000 CHINA China Agricultural University 4 5,000 CHINA Zhejiang University 5 5,000 BRAZIL Universidade de Sao Paulo 6 5,000 BRAZIL Universidade Estadual de Campinas 7 5,000 BELGIUM Ghent University 8 5,000 USA Cornell University 9 5,000 USA University of Massachusetts Amherst 10 5,000 USA University of California Davis 11 5,000 CHINA Northwest A&F University - China 12 5,000 MALAYSIA Universiti Putra Malaysia 13 5,000 IRELAND University College Dublin 14 5,000 PORTUGAL Universidade do Porto 15 5,000 CANADA University of Guelph 16 5,000 CHINA Nanjing Agricultural University 17 5,000 DENMARK University of Copenhagen 18 5,000 BELGIUM KU Leuven 19 5,000 CHINA Nanchang University 20 5,000 ITALY University of Naples Federico II 21 5,000 AUSTRALIA University of Queensland 22 5,000 USA Michigan State University 23 5,000 USA Washington State University 24 5,000 ITALY University of Bologna 25 5,000 USA Rutgers State University New Brunswick 26 5,000 INDIA Indian Institute of Technology System (IIT System) 27 5,000 USA University of Illinois Urbana-Champaign 28 5,000 ITALY University of Milan 29 5,000 USA Ohio State University 30 5,000 USA University of Florida 31 5,000 SPAIN University of Valencia 32 5,000 IRELAND -

Special Lecture Special Lecture 1 July 18 (Wed.) 9:00 - 9:55 Room 1 Program: Program
Special Lecture Special Lecture 1 July 18 (Wed.) 9:00 - 9:55 Room 1 Program: Program: SL1 Toxicity testing in the 21st century—a vision and a strategy Special Lecture Daniel ACOSTA, Jr. Winkle College of Pharmacy, University of Cincinnati, USA Chairperson: Jun KANNO (Division of Cellular & Molecular Toxicology, Biological Safety Research Center, National Institute of Health Sciences, Japan) Special Lecture 2 July 18 (Wed.) 10:00 - 10:55 Room 1 SL2 Cellular adaptive response to environmental toxicants and other noxious stimuli Young-Joon SURH Tumor Microenvironment Global Core Research Center and WCU, Department of Molecular Medicine and Biopharmaceutical Sciences, College of Pharmacy, Seoul National University, Korea Chairperson: Yoshito KUMAGAI (Environmental Medicine Section, Faculty of Medicine, University of Tsukuba, Japan) Special Lecture 3 July 18 (Wed.) 11:00 - 11:55 Room 1 SL3 Molecular basis of Keap1-Nrf2 system function 1 1 1 2 Masayuki YAMAMOTO , Keiko TAGUCHI , Takafumi SUZUKI , Hozumi MOTOHASHI 1Department of Medical Biochemistry, Graduate School of Medicine, Tohoku University, Japan, 2Center for Radioisotope Sciences, Graduate School of Medicine, Tohoku University, Japan Chairperson: Jin-Ho CHUNG (College of Pharmacy, Seoul National University, Korea) Special Lecture 4 July 18 (Wed.) 15:30 - 16:25 Room 1 SL4 Fifty years after the discovery of cytochrome P450: what do we really know about the positive and negative roles in toxicology & health issues? Frederick Peter GUENGERICH Department of Biochemistry, Vanderbilt University School of Medicine, USA Chairperson: Malyn CHULASIRI (Faculty of Pharmacy, Mahidol University, Thailand) Sendai International Center, Sendai, Japan 27 Educational Lecture Educational Lecture 1 July 19 (Thu.) 9:00 - 9:55 Room 1 Educational Lecture Educational EL1 Current understanding of perfluoroalkyl acid toxicology Program: Program: Christopher Si-Lung LAU Toxicity Assessment Division, National Health and Environmental Effects Research Laboratory, Office of Research and Development, U.S. -

21St Century Learning Space Classroom Design in Higher Education: 3D Walkthrough
14th Asian University Presidents’ Forum Hosted by Guangdong University of Foreign Studies Guangzhou, China Dates November 5th ~ November 8th, 2015 Venue Guangdong University of Foreign Studies: North Campus International Academic Exchange Center of GDUFS (Easeland Hotel) Main Theme Asian Higher Education Connectivity: Vision, Process and Approach Sub-Themes Status Quo, Prospects of and Barriers to Asian Higher Education Connectivity and Solutions New Tech: Opportunities and Challenges of Asian Higher Education Connectivity The Roadmap of Asian Higher Education Connectivity Belt and Road Initiative, Interconnectivity and International Education Cooperation Joint Declaration Guangzhou Statement of 2015 Asian University Presidents’ Forum I TABLE OF CONTENTS SCHEDULE OF EVENTS .......................................................................................... 1 WELCOME SPEECH ................................................................................................. 9 DR. SUI GUANGJUN.............................................................................................. 9 CPC Secretary & Chairman of University Administrative Council, Guangdong University of Foreign Studies, China ................................................................... 9 PROMOTING INTERCONNECTIVITY FOR THE FUTURE OF ASIAN HIGHER EDUCATION ................................................................................. 13 DR. ZHONG WEIHE ........................................................................................... 13 President, -

My 45 Years of Chemistry Memoirs of Takeshi Oka (2013 J
Special Issue Preface pubs.acs.org/JPCA My 45 Years of Astrochemistry: Memoirs of Takeshi Oka 1. NH3: CHARLIE TOWNES, KOICHI SHIMODA, JIM theory of vibration−rotation interactions was originated by WATSON Edward Teller, founded by Bright Wilson, and completed by Jim. His 1968 paper on the simplification of the Wilson− I decided to devote my research career to studies of interstellar Howard Hamiltonian is the most beautiful paper on theoretical molecules on December 17, 1968. On that day I read in the spectroscopy by any of my contemporaries.6 Sussex Library of the National Research Council of Canada Following NH ,HOandHCO were discovered in (NRC) in Ottawa the December 16 issue of Physical Review 3 2 2 interstellar clouds. They each appeared in a most dramatic Letters, in which the discovery of interstellar NH3 through radio − way in the centimeter region NH3 in emission from emission between inversion levels of the J=Kmetastable NH3 1 metastable levels, H O as an intense maser with a spectacular was reported by the group of Charlie Townes. Totally unaware 2 velocity profile, and H CO in strong absorption of the 2.73 K that such work had been underway, I was thunderstruck. The 2 cosmic background radiation. All of those manifestations of discovery revealed rich chemistry in interstellar space and extraordinary molecular distributions added fuel to my initiated the concept of “Molecular Clouds” with unexpected fascination with interstellar molecules since each of them is high density, which was to affect astronomy in a profound way. the result of the subtle interplay of collisional and radiative I also had my personal reason to get excited. -

CONICYT Ranking Por Disciplina > Sub-Área OECD (Académicas) Comisión Nacional De Investigación 1
CONICYT Ranking por Disciplina > Sub-área OECD (Académicas) Comisión Nacional de Investigación 1. Ciencias Naturales > 1.4 Ciencias Químicas Científica y Tecnológica PAÍS INSTITUCIÓN RANKING PUNTAJE Nanyang Technological University & National Institute of Education SINGAPORE 1 5,000 (NIE) Singapore CHINA Tsinghua University 2 5,000 INDIA Indian Institute of Technology System (IIT System) 3 5,000 CHINA University of Science & Technology of China 4 5,000 CHINA Zhejiang University 5 5,000 CHINA Peking University 6 5,000 USA University of California Berkeley 7 5,000 CHINA Jilin University 8 5,000 USA Massachusetts Institute of Technology (MIT) 9 5,000 CHINA South China University of Technology 10 5,000 CHINA Suzhou University 11 5,000 USA Stanford University 12 5,000 SINGAPORE National University of Singapore 13 5,000 CHINA Nanjing University 14 5,000 CHINA Fudan University 15 5,000 SAUDI ARABIA King Abdulaziz University 16 5,000 USA University of Chicago 17 5,000 CHINA Nankai University 18 5,000 JAPAN Kyoto University 19 5,000 CHINA Shanghai Jiao Tong University 20 5,000 SWITZERLAND Ecole Polytechnique Federale de Lausanne 21 5,000 CHINA Tianjin University 22 5,000 USA Georgia Institute of Technology 23 5,000 CHINA Sichuan University 24 5,000 UNITED KINGDOM University of Cambridge 25 5,000 USA Northwestern University 26 5,000 SOUTH KOREA University of Science & Technology (UST) 27 5,000 JAPAN University of Tokyo 28 5,000 UNITED KINGDOM University of Oxford 29 5,000 CHINA Huazhong University of Science & Technology 30 5,000 CHINA Wuhan University -
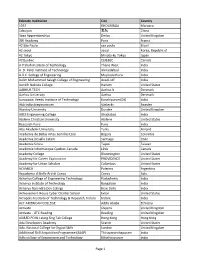
List AWS Educate Institutions
Educate Institution City Country 1337 KHOURIBGA Morocco 1daoyun 无锡 China 3aaa Apprenticeships Derby United Kingdom 3W Academy Paris France 42 São Paulo sao paulo Brazil 42 seoul seoul Korea, Republic of 42 Tokyo Minato-ku Tokyo Japan 42Quebec QUEBEC Canada A P Shah Institute of Technology Thane West India A. D. Patel Institute of Technology Ahmedabad India A.V.C. College of Engineering Mayiladuthurai India Aalim Muhammed Salegh College of Engineering Avadi-IAF India Aaniiih Nakoda College Harlem United States AARHUS TECH Aarhus N Denmark Aarhus University Aarhus Denmark Aarupadai Veedu Institute of Technology Kanchipuram(Dt) India Abb Industrigymansium Västerås Sweden Abertay University Dundee United Kingdom ABES Engineering College Ghaziabad India Abilene Christian University Abilene United States Research Pune Pune India Abo Akademi University Turku Finland Academia de Bellas Artes Semillas Ltda Bogota Colombia Academia Desafio Latam Santiago Chile Academia Sinica Taipei Taiwan Academie Informatique Quebec-Canada Lévis Canada Academy College Bloomington United States Academy for Career Exploration PROVIDENCE United States Academy for Urban Scholars Columbus United States ACAMICA Palermo Argentina Accademia di Belle Arti di Cuneo Cuneo Italy Achariya College of Engineering Technology Puducherry India Acharya Institute of Technology Bangalore India Acharya Narendra Dev College New Delhi India Achievement House Cyber Charter School Exton United States Acropolis Institute of Technology & Research, Indore Indore India ACT AMERICAN COLLEGE -

FORMATO PDF Ranking Instituciones Acadã©Micas Por Sub Ã
Ranking Instituciones Académicas por sub área OCDE 2020 2. Ingeniería y Tecnología > 2.08 Biotecnología Medioambiental PAÍS INSTITUCIÓN RANKING PUNTAJE USA Harvard University 1 5,000 USA Massachusetts Institute of Technology (MIT) 2 5,000 USA Stanford University 3 5,000 USA University of Pennsylvania 4 5,000 UNITED KINGDOM University College London 5 5,000 UNITED KINGDOM University of Cambridge 6 5,000 USA University of California San Diego 7 5,000 USA University of California Berkeley 8 5,000 DENMARK Technical University of Denmark 9 5,000 CHINA Zhejiang University 10 5,000 USA University of Washington Seattle 11 5,000 USA Cornell University 12 5,000 USA Baylor College of Medicine 13 5,000 USA Johns Hopkins University 14 5,000 USA University of California Los Angeles 15 5,000 CANADA University of Toronto 16 5,000 BELGIUM Ghent University 17 5,000 AUSTRALIA University of Queensland 18 5,000 SOUTH KOREA Seoul National University (SNU) 19 5,000 DENMARK University of Copenhagen 20 5,000 CHINA Shanghai Jiao Tong University 21 5,000 USA University of Minnesota Twin Cities 22 5,000 CHINA Harbin Institute of Technology 23 5,000 USA Ohio State University 24 5,000 USA University of Florida 25 5,000 SINGAPORE National University of Singapore 26 5,000 NETHERLANDS Wageningen University & Research 27 5,000 GERMANY RWTH Aachen University 28 5,000 SAUDI ARABIA King Abdulaziz University 29 5,000 BRAZIL Universidade de Sao Paulo 30 5,000 USA University of California Davis 31 5,000 USA University of California San Francisco 32 5,000 CHINA Tsinghua University 33 5,000 CHINA Jiangnan University 34 5,000 UNITED KINGDOM University of Oxford 35 5,000 AUSTRALIA University of Melbourne 36 5,000 GERMANY Karlsruhe Institute of Technology 37 5,000 UNITED KINGDOM University of Edinburgh 38 5,000 CHINA Shandong University 39 5,000 UNITED KINGDOM Imperial College London 40 5,000 USA University of Illinois Urbana-Champaign 41 5,000 USA University of Wisconsin Madison 42 5,000 USA University of Michigan 43 5,000 USA Harvard T.H. -

2008 2Nd Annual Dasari Lecture Takeshi Oka, University Of
2008 2nd Annual Dasari Lecture Takeshi Oka, University of Chicago “H3+ in the central molecular zone of the Galactic center” Takeshi Oka is the world leader in the infrared spectroscopy of molecular ions. Born in Tokyo in 1932, Professor Oka received his Ph.D. degree at the University of Tokyo in 1960. He was at the National Research Council of Canada from 1963 to 1981 and in 1975, he joined the prestigious Herzberg Institute of Astrophysics in Ottawa, working with the Nobel Laureate Gerhard Herzberg, where, in 1980, he measured the fundamental spectrum of the H3+ molecule. Shortly after his discovery of the H3+ spectrum, Professor Oka moved to the University of Chicago as Professor of Chemistry and Astronomy and Astrophysics to establish his own laboratory, affectionately known as the Oka Ion Factory. He was appointed Robert A. Millikan Distinguished Service Professor in 1989, and Emeritus in 2003. The unifying theme of Professor Oka’s studies is the understanding of the quantum mechanics and dynamics of fundamental molecular ions and their behavior in both laboratory and astronomical environments. His work has ranged over many oxygen, nitrogen and carbon-bearing molecular ions, but his name will always be especially associated with H3+. As the simplest of all poly-atomic molecular ions, H3+ has proved a test-bed for chemical and spectroscopic studies, in the laboratory and in theory, which Professor Oka has spearheaded. H3+ has also proved a starting point for the techniques, experimental and theoretical, used by the Oka Ion Factory to measure and interpret dozens of molecular ion spectra over the past two decades.