Transferrin Binding Protein B Structure, Function, and Export in Neisseria Gonorrhoeae
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Hgba Locus Encoding a Hemoglobin Receptor from Haemophilus Ducreyi
INFECTION AND IMMUNITY, June 1995, p. 2194–2200 Vol. 63, No. 6 0019-9567/95/$04.0010 Copyright q 1995, American Society for Microbiology Characterization of the hgbA Locus Encoding a Hemoglobin Receptor from Haemophilus ducreyi 1 1 2 CHRISTOPHER ELKINS, * CHING-JU CHEN, AND CHRISTOPHER E. THOMAS Departments of Medicine1 and of Microbiology and Immunology,2 School of Medicine, University of North Carolina, Chapel Hill, North Carolina 27599 Received 1 December 1994/Returned for modification 8 February 1995/Accepted 22 March 1995 Haemophilus ducreyi can bind hemoglobin and use it as a source of heme, for which it has an obligate requirement. We previously identified and purified HgbA, a hemoglobin-binding outer membrane protein from H. ducreyi. In this report, we describe the molecular cloning, expression, DNA sequence, and mutagenesis of the structural gene for HgbA, hgbA. H. ducreyi and recombinant Escherichia coli expressing hgbA bound [125I]he- moglobin, establishing HgbA as a receptor. Insertions or deletions in the cloned hgbA gene abolished expres- sion of HgbA and hemoglobin binding in E. coli. Mutagenesis of H. ducreyi by allelic exchange of insertions into hgbA abolished its ability to bind [125I]hemoglobin or utilize hemoglobin as a source of heme. The deduced protein sequence was similar to those of the TonB-dependent family of outer membrane receptors. The most similar member was HutA (heme receptor) from Vibrio cholerae. Tbp1 and Lbp1 (transferrin and lactoferrin receptors, respectively, from pathogenic Neisseria spp.) also showed very significant homology. Thus, by characterizing the hgbA locus, this work elucidates a potentially important role of HgbA in obtaining heme and/or iron from the host. -

The Role of Electrostatics in Siderophore Recognition by the Immunoprotein Siderocalin1 Trisha M
Published on Web 11/19/2008 The Role of Electrostatics in Siderophore Recognition by the Immunoprotein Siderocalin1 Trisha M. Hoette,† Rebecca J. Abergel,† Jide Xu,† Roland K. Strong,‡ and Kenneth N. Raymond*,† Department of Chemistry, UniVersity of California, Berkeley, California 94720-1460, and DiVision of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109 Received September 19, 2008; E-mail: [email protected] Abstract: Iron is required for virulence of most bacterial pathogens, many of which rely on siderophores, small-molecule chelators, to scavenge iron in mammalian hosts. As an immune response, the human protein Siderocalin binds both apo and ferric siderophores in order to intercept delivery of iron to the bacterium, impeding virulence. The introduction of steric clashes into the siderophore structure is an important mechanism of evading sequestration. However, in the absence of steric incompatibilities, electrostatic interactions determine siderophore strength of binding by Siderocalin. By using a series of isosteric enterobactin analogues, the contribution of electrostatic interactions, including both charge-charge and cation-π, to the recognition of 2,3-catecholate siderophores has been deconvoluted. The analogues used in the study incorporate a systematic combination of 2,3-catecholamide (CAM) and N-hydroxypyridinonate (1,2-HOPO) binding units on a tris(2-aminoethyl)amine (tren) backbone, [tren(CAM)m(1,2-HOPO)n, where m ) 0, 1, 2, or 3 and n ) 3 - m]. The shape complementarity of the synthetic analogue series was determined through small-molecule crystallography, and the binding interactions were investigated through a fluorescence-based binding assay. These results were modeled and correlated through ab initio calculations of the electrostatic properties of the binding units. -

Predicting Weakly Stable Regions, Oligomerization State, and Protein–Protein Interfaces in Transmembrane Domains of Outer Membrane Proteins
Predicting weakly stable regions, oligomerization state, and protein–protein interfaces in transmembrane domains of outer membrane proteins Hammad Naveed, Ronald Jackups Jr., and Jie Liang1 Department of Bioengineering, University of Illinois, Chicago, IL 60607 Edited by William F. DeGrado, University of Pennsylvania School of Medicine, Philadelphia, PA, and approved June 11, 2009 (received for review February 26, 2009) Although the structures of many -barrel membrane proteins are membrane proteins, such as voltage-sensing (11), flux control of available, our knowledge of the principles that govern their ener- metabolites, and ion-sensing (9). getics and oligomerization states is incomplete. Here we describe Computational studies have also contributed much to the a computational method to study the transmembrane (TM) do- understanding of -barrel membrane proteins, including the mains of -barrel membrane proteins. Our method is based on a identification of -barrel membrane proteins from sequences of physical interaction model, a simplified conformational space for many microbial genomes, the prediction of their topological efficient enumeration, and an empirical potential function from a orientations, and the characterization of their structural features detailed combinatorial analysis. Using this method, we can identify (12, 13). In addition, numerous spatial and sequence motifs and weakly stable regions in the TM domain, which are found to be ensemble properties of the TM domains have also been studied important structural determinants for -barrel membrane pro- (13–18). As models and algorithms improve, it is natural to ask teins. By calculating the melting temperatures of the TM strands, whether computational studies can reveal further insight on the our method can also assess the stability of -barrel membrane structural organization of -barrel membrane proteins. -

Engineering Antimicrobial Probiotics for the Treatment of Vancomycin-Resistant Enterococcus
Engineering Antimicrobial Probiotics for the Treatment of Vancomycin-Resistant Enterococcus A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY KATHRYN GELDART IN PARTIAL FULLFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY ADVISOR: YIANNIS N. KAZNESSIS December, 2016 © Kathryn Geldart 2016 ACKNOWLEDGEMENTS I am deeply grateful to my Ph.D. advisor, Yiannis Kaznessis for his guidance, encouragement, and unwavering faith in my abilities throughout my time as his graduate student. His optimism and enthusiasm towards this project provided a constant fuel of inspiration, both in times of success and frustration. The trust he instilled in me gave me confidence and freedom to explore unfamiliar techniques and topics. We now know more about E. faecium resistance than either one of us originally intended. Once again, thank you Yiannis, for everything. I must also thank our collaborator, Gary Dunny, for his support and advice on bacterial techniques used throughout this project. Gary’s expertise on Enterococcus has played a key role in this project and I am incredibly grateful for the time he has taken for all of our discussions. I thank the members of Gary’s lab, especially PostDocs Dawn Manias, Jennifer Dale, and Yuqing Chen who taught me numerous techniques that have been fundamental to this work. I also thank our other collaborators, Nita Salzman and her PostDoc Sushma Kommineni for their expert advice on Enterococcus in in vivo settings and for the vast efforts they have put into the mouse trials. I thank my current lab mates Brittany Forkus and Seth Ritter for your invaluable input, support, and daily comic relief. -

Antibacterial Prodrugs to Overcome Bacterial Resistance
molecules Review Antibacterial Prodrugs to Overcome Bacterial Resistance Buthaina Jubeh , Zeinab Breijyeh and Rafik Karaman * Pharmaceutical Sciences Department, Faculty of Pharmacy, Al-Quds University, Jerusalem P.O. Box 20002, Palestine; [email protected] (B.J.); [email protected] (Z.B.) * Correspondence: [email protected] or rkaraman@staff.alquds.edu Academic Editor: Helen Osborn Received: 10 March 2020; Accepted: 26 March 2020; Published: 28 March 2020 Abstract: Bacterial resistance to present antibiotics is emerging at a high pace that makes the development of new treatments a must. At the same time, the development of novel antibiotics for resistant bacteria is a slow-paced process. Amid the massive need for new drug treatments to combat resistance, time and effort preserving approaches, like the prodrug approach, are most needed. Prodrugs are pharmacologically inactive entities of active drugs that undergo biotransformation before eliciting their pharmacological effects. A prodrug strategy can be used to revive drugs discarded due to a lack of appropriate pharmacokinetic and drug-like properties, or high host toxicity. A special advantage of the use of the prodrug approach in the era of bacterial resistance is targeting resistant bacteria by developing prodrugs that require bacterium-specific enzymes to release the active drug. In this article, we review the up-to-date implementation of prodrugs to develop medications that are active against drug-resistant bacteria. Keywords: prodrugs; biotransformation; targeting; β-lactam antibiotics; β-lactamases; pathogens; resistance 1. Introduction Nowadays, the issue of pathogens resistant to drugs and the urgent need for new compounds that are capable of eradicating these pathogens are well known and understood. -

And Iron-Binding Eukaryotic Proteins (Bottom: Molecular Mass 80, 000 - 500,000 D)
Iron and the Pathogenicity of Bacteria Phillip E. Klebba, Ph. D. and Salete M. C. Newton, Ph. D. I. Gram-negative bacterial iron uptake. We are studying the ability of pathogenic bacteria to obtain the element iron (Fe) in human and animal hosts. This research spans several decades, which may be briefly summarized with a few statements. First, not just microorganisms, but essentially all organisms, require iron for a variety of metabolic processes, including energy generation by cytochrome-containing proteins, DNA synthesis, as a cofactor in metabolic enzymes, and for detoxification of reactive oxygen species. Secondly, humans and animals sequester iron within the body, in forms like transferrin, lactoferrin and ferritin, as a means of defense against prokaryotic infection, but microorganisms synthesize and secrete small organic molecules called siderophores that actively chelate iron and remove it from eukaryotic iron-binding proteins (Fig. 1). Furthermore, some bacterial pathogens may directly utilize the iron in transferrin or lactoferrin. Thus on the molecular level, iron is a valuable commodity that is a key element of bacterial pathogenesis. But unfortunately, the process of iron acquisition is not well understood in either Gram-positive or Gram-negative bacteria. Figure 1. Structure of iron-binding microbial siderophores (top; molecular mass 700 - 1000 D) and iron-binding eukaryotic proteins (bottom: molecular mass 80, 000 - 500,000 D). For comparison, the actual size and structure of ferric enterobactin is shown next to that of ferritin. Regarding Gram-negative organisms, our experiments focus on the uptake of iron through the outer membrane (OM) of Escherichia coli (Fig. 2) , a prototypic bacterium that is like many other pathogens, including Salmonella typhi (typhoid fever), Vibrio cholerae (cholera), Shigella dysenteria (dysentery), Neisseria meningitidis (meningitis) and Yersinia pestis (plague). -

Mechanisms of the Antibacterial Activity of Lactoferrin and Lactoferrin-Derived Peptides
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/268421952 MECHANISMS OF THE ANTIBACTERIAL ACTIVITY OF LACTOFERRIN AND LACTOFERRIN-DERIVED PEPTIDES Article CITATIONS READS 0 408 3 authors: Anca Roseanu Maria Damian Institutul de Biochimie Cantacuzino Institute 39 PUBLICATIONS 711 CITATIONS 50 PUBLICATIONS 308 CITATIONS SEE PROFILE SEE PROFILE Robert W Evans 129 PUBLICATIONS 4,253 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: CEEX 47 - Programme of Romanian Ministry of Education and Research View project Modeling and Simulation of Proteins Structure and Interactions View project All content following this page was uploaded by Anca Roseanu on 15 April 2015. The user has requested enhancement of the downloaded file. MECHANISMS OF THE ANTIBACTERIAL ACTIVITY OF LACTOFERRIN AND LACTOFERRIN-DERIVED PEPTIDES ANCA ROŞEANU1*, MARIA DAMIAN2, ROBERT W. EVANS3 1Institute of Biochemistry of the Romanian Academy, Splaiul Independenţei 296, Bucharest, Romania 2“I. Cantacuzino” National Institute of Research-Development for Microbiology and Immunology, Splaiul Independenţei 103, Bucharest, Romania 3Metalloprotein Research Group, Division of Biosciences, Brunel University, Uxbridge, Middlesex, UB8 3PH, UK (Received October 15, 2010) Lactoferrin (Lf), a member of the transferrin family of iron-binding proteins, is now known to have a number of properties, including antibacterial activity towards a broad spectrum of Gram-negative and Gram-positive bacteria. The mechanism of the antibacterial activity of Lf is complex and involves beside iron-chelation, direct action on bacteria and/or the activation of the immune system. Lactoferricin (Lfcin) and other peptides derived from Lf or Lfcin are more potent antibacterial agents, a property exhibited by interaction with and penetration of bacterial membrane. -

Ferric Enterobactin Binding and Utilization by Neisseria Gonorrhoeae SUSAN D
JOURNAL OF BACTERIOLOGY, May 1999, p. 2895–2901 Vol. 181, No. 9 0021-9193/99/$04.0010 Copyright © 1999, American Society for Microbiology. All Rights Reserved. Ferric Enterobactin Binding and Utilization by Neisseria gonorrhoeae SUSAN D. BIEGEL CARSON,1 PHILIP E. KLEBBA,2 SALETE M. C. NEWTON,2 1,3 AND P. FREDERICK SPARLING * Department of Microbiology and Immunology1 and Department of Medicine,3 University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, and Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 730192 Received 23 December 1998/Accepted 19 February 1999 FetA, formerly designated FrpB, an iron-regulated, 76-kDa neisserial outer membrane protein, shows se- quence homology to the TonB-dependent family of receptors that transport iron into gram-negative bacteria. Although FetA is commonly expressed by most neisserial strains and is a potential vaccine candidate for both Neisseria gonorrhoeae and Neisseria meningitidis, its function in cell physiology was previously undefined. We now report that FetA functions as an enterobactin receptor. N. gonorrhoeae FA1090 utilized ferric enterobactin as the sole iron source when supplied with ferric enterobactin at approximately 10 mM, but growth stimulation was abolished when an omega (V) cassette was inserted within fetA or when tonB was insertionally interrupted. 59 m FA1090 FetA specifically bound Fe-enterobactin, with a Kd of approximately 5 M. Monoclonal antibodies raised against the Escherichia coli enterobactin receptor, FepA, recognized FetA in Western blots, and amino acid sequence comparisons revealed that residues previously implicated in ferric enterobactin binding by FepA were partially conserved in FetA. An open reading frame downstream of fetA, designated fetB, predicted a protein with sequence similarity to the family of periplasmic binding proteins necessary for transporting sidero- phores through the periplasmic space of gram-negative bacteria. -
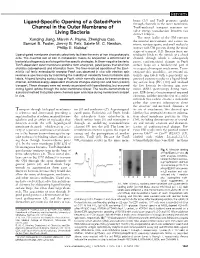
Ligand-Specific Opening of a Gated
REPORTS brane (11) and TonB promotes uptake Ligand-Specific Opening of a Gated-Porin through channels in the outer membrane, Channel in the Outer Membrane of TonB-mediated transport reactions in- volve energy transduction between two Living Bacteria distinct bilayers. The outer leaflet of the OM contacts Xunqing Jiang, Marvin A. Payne, Zhenghua Cao, the external environment, and various nu- Samuel B. Foster, Jimmy B. Feix, Salete M. C. Newton, trients, noxious agents, and small molecules Phillip E. Klebba* interact with OM proteins during the initial stages of transport (12). Because ferric en- Ligand-gated membrane channels selectively facilitate the entry of iron into prokaryotic terobactin binds to the outside of a closed cells. The essential role of iron in metabolism makes its acquisition a determinant of channel, through which it subsequently bacterial pathogenesis and a target for therapeutic strategies. In Gram-negative bacteria, passes, conformational changes in FepA TonB-dependent outer membrane proteins form energized, gated pores that bind iron surface loops are a fundamental part of chelates (siderophores) and internalize them. The time-resolved operation of the Esch- its suspected transport mechanism. To in- erichia coli ferric enterobactin receptor FepA was observed in vivo with electron spin vestigate this possibility, we reacted ni- resonance spectroscopy by monitoring the mobility of covalently bound nitroxide spin troxide spin labels with a genetically en- labels. A ligand-binding surface loop of FepA, which normally closes its transmembrane gineered cysteine residue in a ligand-bind- channel, exhibited energy-dependent structural changes during iron and toxin (colicin) ing surface loop [PL5 (13)] and analyzed transport. -
Expression of Iron, the Salmochelin Siderophore Receptor, Requires
Expression of IroN, the salmochelin siderophore receptor, requires mRNA activation by RyhB small RNA homologues Roberto Balbontín, Nicolás Villagra, Maria Pardos de la Gándara, Guido Mora, Nara Figueroa-Bossi, Lionello Bossi To cite this version: Roberto Balbontín, Nicolás Villagra, Maria Pardos de la Gándara, Guido Mora, Nara Figueroa- Bossi, et al.. Expression of IroN, the salmochelin siderophore receptor, requires mRNA activation by RyhB small RNA homologues. Molecular Microbiology, Wiley, 2016, 100 (1), pp.139 - 155. 10.1111/mmi.13307. hal-01412678 HAL Id: hal-01412678 https://hal.archives-ouvertes.fr/hal-01412678 Submitted on 8 Dec 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Expression of IroN, the salmochelin siderophore receptor, requires mRNA activation by RyhB small RNA homologues Roberto Balbontın,1† Nicolas Villagra,2 to the polychromatic landscape of sRNA-mediated Maria Pardos de la Gandara, 1‡ Guido Mora,2 regulation. Nara Figueroa-Bossi1* and Lionello Bossi1 1Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Universite Paris-Sud, Universite Paris- Introduction Saclay, 91198 Gif-sur-Yvette, France. Rewiring of gene expression networks in response to 2Laboratorio de Patogenesis Molecular y chemical or physical cues is central to the ability of bac- Antimicrobianos, Facultad de Medicina, Universidad terial pathogens to adapt to environmental changes. -

Iron-Withholding Strategy in Innate Immunity
ARTICLE IN PRESS Immunobiology 211 (2006) 295–314 www.elsevier.de/imbio REVIEW Iron-withholding strategy in innate immunity Sek Tong Onga, Jason Zhe Shan Hob, Bow Hoc,1, Jeak Ling Dinga,1,Ã aDepartment of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543 bFaculty of Medicine, Imperial College London, South Kensington, London SW7 2AZ, UK cDepartment of Microbiology, National University of Singapore, 5 Science Drive 2, Singapore 117597 Received 13 January 2006; accepted 14 February 2006 Abstract The knowledge of how organisms fight infections has largely been built upon the ability of host innate immune molecules to recognize microbial determinants. Although of overwhelming importance, pathogen recognition is but only one of the facets of innate immunity. A primitive yet effective antimicrobial mechanism which operates by depriving microbial organisms of their nutrients has been brought into the forefront of innate immunity once again. Such a tactic is commonly referred to as the iron-withholding strategy of innate immunity. In this review, we introduce various vertebrate iron-binding proteins and their invertebrate homologues, so as to impress upon readers an obscured arm of innate immune defense. An excellent comprehension of the mechanics of innate immunity paves the way for the possibility that novel antimicrobial therapeutics may emerge one day to overcome the prevalent antibiotic resistance in bacteria. r 2006 Elsevier GmbH. All rights reserved. Keywords: Innate immunity; Iron sequestration; Ferritin; Hepcidin; Lipocalin; Nramp; Transferrin Contents Introduction . 296 Iron as a double-edged sword in biological systems . 296 How is iron involved in innate immunity? . 297 Advances in vertebrate host innate immune defense: the iron-withholding strategy . -

Cloning, Mapping and Identification of the Aeromonas Salmonicida Fsta
魚 病 研 究 Fish Pathology,33(4),239-246,1998.10 Cloning,Mapping and Identificationof theAeromonas salmonicidafstA,fepA and irpAGenes, Encoding the 86,82 and 74 kDa Iron-regulated Outer Membrane Proteins,Respectively R.J. Collighan, A. J. Bennett and G. Coleman Department of Biochemistry,NottinghamUniversity Medical School,Queen'sMedical Centre, Clifton Boulevard, Nottingham,NG7 2UH,U.K. (Received February 23,1998) Three iromps(iron-regulatedouter membrane proteins)ofAeromonas salmonicidawere identifiedby the use of specificantibodies together with Southernhybridization analysis and limitednucleotide sequencing of their genes. The resultsof theseexperiments together with a searchof the internationaldatabase for homologous sequencesled to theiridentification as follows: 86 kDa iromp(FstA)as a Vibrioanguillarum Fat A homologue 82 kDa iromp(FepA)as an Escherichiacoli FepA homologue 74kDa iromp(IrpA)as an Escherichiacoli Cir homologue Key words : gene cloning,gene mapping,Aeromonas salmonicida,iron-regulated outer membrane protein,iromp Pathogenic bacteria require iron for growth in a host such are considered as potential vaccine components where the only source of this element is tightly com (Banerjee-Bhatnagar and Frasch, 1990; Robinson and plexed in a number of iron-binding proteins such as Melling, 1993). An earlier study by Chart and Trust haemoglobin, transferrin and lactoferrin. Two major (1983) detected an inducible siderophore-mediatedtrans mechanisms for acquiring iron from the host proteins port system in the fish pathogen,Aeromonas salmonicida, have been described. The first, present in Escherichia the causative agent of furunculosis in salmonid fish. coli requires the formation of soluble factors with a high Hirst et al. (1991) later identified the siderophore as a affinity for iron such that they are able to remove it from 2,3 diphenol-catechol derivative.