Chromatin Accessibility Underlies Synthetic Lethality of SWI/SNF
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ARID1A Protein Expression Is Retained in Ovarian Endometriosis
www.nature.com/scientificreports OPEN ARID1A protein expression is retained in ovarian endometriosis with ARID1A loss‑of‑function mutations: implication for the two‑hit hypothesis Nozomi Yachida1, Kosuke Yoshihara1*, Kazuaki Suda1, Hirofumi Nakaoka2,3, Haruka Ueda1, Kentaro Sugino1, Manako Yamaguchi1, Yutaro Mori1, Kaoru Yamawaki1, Ryo Tamura1, Tatsuya Ishiguro1, Masanori Isobe1, Teiichi Motoyama4, Ituro Inoue2 & Takayuki Enomoto1 ARID1A loss‑of‑function mutation accompanied by a loss of ARID1A protein expression is considered one of the most important driver events in endometriosis‑associated ovarian cancer. Although our recent genomic study clarifed that ARID1A loss‑of‑function mutations were detected in 13% of ovarian endometriosis, an association between the ARID1A mutation status and ARID1A protein expression in ovarian endometriosis remains unclear. We performed immunohistochemical staining for ARID1A in 78 ovarian endometriosis samples and 99 clear cell carcinoma samples. We revealed that not only 70 endometriosis samples without ARID1A mutations but also eight endometriosis samples with ARID1A loss‑of‑function mutations retained ARID1A protein expression. On the other hand, most of clear cell carcinomas with ARID1A loss‑of‑function mutations showed a loss of ARID1A protein expression. In particular, clear cell carcinoma samples which harbor multiple ARID1A loss‑of‑ function mutations or both a single ARID1A loss‑of‑function mutation and ARID1A allelic imbalance lost ARID1A protein expression. However, ARID1A protein expression was retained in seven clear cell carcinomas with ARID1A loss‑of‑function mutations. These results suggest that a single ARID1A loss‑of‑function mutation is insufcient for ARID1A loss in ovarian endometriosis and some clear cell carcinoma. Further driver events may be needed for the malignant transformation of ovarian endometriosis with ARID1A loss‑of‑function mutations. -

Avian Binocularity and Adaptation to Nocturnal Environments: Genomic Insights Froma Highly Derived Visual Phenotype Rui Borges Universidade Do Porto - Portugal
Nova Southeastern University NSUWorks Biology Faculty Articles Department of Biological Sciences 8-22-2019 Avian Binocularity and Adaptation to Nocturnal Environments: Genomic Insights froma Highly Derived Visual Phenotype Rui Borges Universidade do Porto - Portugal Joao Fonseca Universidade do Porto - Portugal Cidalia Gomes Universidade do Porto - Portugal Warren E. Johnson Smithsonian Institution Stephen James O'Brien St. Petersburg State University - Russia; Nova Southeastern University, [email protected] See next page for additional authors Follow this and additional works at: https://nsuworks.nova.edu/cnso_bio_facarticles Part of the Biology Commons NSUWorks Citation Borges, Rui; Joao Fonseca; Cidalia Gomes; Warren E. Johnson; Stephen James O'Brien; Guojie Zhang; M. Thomas P. Gilbert; Erich D. Jarvis; and Agostinho Antunes. 2019. "Avian Binocularity and Adaptation to Nocturnal Environments: Genomic Insights froma Highly Derived Visual Phenotype." Genome Biology and Evolution 11, (8): 2244-2255. doi:10.1093/gbe/evz111. This Article is brought to you for free and open access by the Department of Biological Sciences at NSUWorks. It has been accepted for inclusion in Biology Faculty Articles by an authorized administrator of NSUWorks. For more information, please contact [email protected]. Authors Rui Borges, Joao Fonseca, Cidalia Gomes, Warren E. Johnson, Stephen James O'Brien, Guojie Zhang, M. Thomas P. Gilbert, Erich D. Jarvis, and Agostinho Antunes This article is available at NSUWorks: https://nsuworks.nova.edu/cnso_bio_facarticles/982 GBE Avian Binocularity and Adaptation to Nocturnal Environments: Genomic Insights from a Highly Derived Visual Downloaded from https://academic.oup.com/gbe/article-abstract/11/8/2244/5544263 by Nova Southeastern University/HPD Library user on 16 September 2019 Phenotype Rui Borges1,2,Joao~ Fonseca1,Cidalia Gomes1, Warren E. -

The Role of Components of the Chromatin Modification Machinery in Carcinogenesis of Clear Cell Carcinoma of the Ovary (Review)
ONCOLOGY LETTERS 2: 591-597, 2011 The role of components of the chromatin modification machinery in carcinogenesis of clear cell carcinoma of the ovary (Review) HIROSHI SHIGETOMI, AKIRA OONOGI, TAIHEI TSUNEMI, YASUHITO TANASE, YOSHIHIKO YAMADA, HIROTAKA KAJIHARA, YORIKO YOSHIZAWA, NAOTO FURUKAWA, SHOJI HARUTA, SHOZO YOSHIDA, TOSHIYUKI SADO, HIDEKAZU OI and HIROSHI KOBAYASHI Department of Obstetrics and Gynecology, Nara Medical University, Nara, Japan Received January 21, 2011; Accepted April 27, 2011 DOI: 10.3892/ol.2011.316 Abstract. Recent data have provided information regarding 6. A marked resemblance between CCC and ccRCC the profiles of clear cell carcinoma of the ovary (CCC) with 7. Conclusions adenine-thymine rich interactive domain 1A (ARID1A) muta- tions. The purpose of this review was to summarize current 1. Introduction knowledge regarding the molecular mechanisms involved in CCC tumorigenesis and to describe the central role played Epithelial ovarian cancer (EOC) is the most lethal gyne- by the aberrant chromatin remodeling. The present article cologic malignancy worldwide. Epidemiology calculations reviews the English-language literature for biochemical of lifetime risk for EOC are that 1 in 55 women is likely to studies on the ARID1A mutation and chromatin remodeling develop EOC during their lifetime (1). Since EOC is more in CCC. ARID1A is responsible for directing the SWI/SNF likely to be advanced stage with unfavorable tumor biology, complex to target promoters and regulates the transcription of there are serious limitations to the surgical and oncological certain genes by altering the chromatin structure around those treatment available. Therefore, it is crucial to determine the genes. The mutation spectrum of ARID1A was enriched for earliest possible diagnosis. -

Acquired Mutations and Transcriptional Remodeling in Long-Term Estrogen-Deprived Locoregional Breast Cancer Recurrences
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.140707; this version posted June 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic remodeling in endocrine resistant local breast cancer recurrences Lee 1 Manuscript Submission Acquired mutations and transcriptional remodeling in long-term estrogen-deprived locoregional breast cancer recurrences An Original Research Article by: Nolan Priedigkeit1,3, Kai Ding2,3,4, William Horne5, Jay K. Kolls5, Tian Du3, Peter C. Lucas3,6, Jens-Uwe Blohmer7, Carsten Denkert8, Anna Machleidt7, Barbara Ingold-Heppner9, Steffi Oesterreich2,3,4, Adrian V. Lee2,3,4,10 1Department of Medicine, Brigham and Women’s Hospital / Harvard Medical School, Boston, Massachusetts 2Department of Pharmacology and Chemical Biology, University of Pittsburgh, Pittsburgh, Pennsylvania 3Women's Cancer Research Center, UPMC Hillman Cancer Center, Pittsburgh, Pennsylvania 4Magee-Women's Research Institute, Magee-Women's Research Hospital of University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania 5Richard King Mellon Foundation Institute for Pediatric Research, UPMC Children's Hospital of Pittsburgh, Pittsburgh, Pennsylvania, USA. 6Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 7Institute of Pathology and Department of Gynecology, Charité University Hospital, Berlin, -

The Expression of Genes Contributing to Pancreatic Adenocarcinoma Progression Is Influenced by the Respective Environment – Sagini Et Al
The expression of genes contributing to pancreatic adenocarcinoma progression is influenced by the respective environment – Sagini et al Supplementary Figure 1: Target genes regulated by TGM2. Figure represents 24 genes regulated by TGM2, which were obtained from Ingenuity Pathway Analysis. As indicated, 9 genes (marked red) are down-regulated by TGM2. On the contrary, 15 genes (marked red) are up-regulated by TGM2. Supplementary Table 1: Functional annotations of genes from Suit2-007 cells growing in pancreatic environment Categoriesa Diseases or p-Valuec Predicted Activation Number of genesf Functions activationd Z-scoree Annotationb Cell movement Cell movement 1,56E-11 increased 2,199 LAMB3, CEACAM6, CCL20, AGR2, MUC1, CXCL1, LAMA3, LCN2, COL17A1, CXCL8, AIF1, MMP7, CEMIP, JUP, SOD2, S100A4, PDGFA, NDRG1, SGK1, IGFBP3, DDR1, IL1A, CDKN1A, NREP, SEMA3E SERPINA3, SDC4, ALPP, CX3CL1, NFKBIA, ANXA3, CDH1, CDCP1, CRYAB, TUBB2B, FOXQ1, SLPI, F3, GRINA, ITGA2, ARPIN/C15orf38- AP3S2, SPTLC1, IL10, TSC22D3, LAMC2, TCAF1, CDH3, MX1, LEP, ZC3H12A, PMP22, IL32, FAM83H, EFNA1, PATJ, CEBPB, SERPINA5, PTK6, EPHB6, JUND, TNFSF14, ERBB3, TNFRSF25, FCAR, CXCL16, HLA-A, CEACAM1, FAT1, AHR, CSF2RA, CLDN7, MAPK13, FERMT1, TCAF2, MST1R, CD99, PTP4A2, PHLDA1, DEFB1, RHOB, TNFSF15, CD44, CSF2, SERPINB5, TGM2, SRC, ITGA6, TNC, HNRNPA2B1, RHOD, SKI, KISS1, TACSTD2, GNAI2, CXCL2, NFKB2, TAGLN2, TNF, CD74, PTPRK, STAT3, ARHGAP21, VEGFA, MYH9, SAA1, F11R, PDCD4, IQGAP1, DCN, MAPK8IP3, STC1, ADAM15, LTBP2, HOOK1, CST3, EPHA1, TIMP2, LPAR2, CORO1A, CLDN3, MYO1C, -

Genome-Wide Crosstalk Between Steroid Receptors in Breast and Prostate Cancers
28 9 Endocrine-Related V Paakinaho and J J Palvimo Steroid receptor crosstalk in 28:9 R231–R250 Cancer cancers REVIEW Genome-wide crosstalk between steroid receptors in breast and prostate cancers Ville Paakinaho and Jorma J Palvimo Institute of Biomedicine, School of Medicine, University of Eastern Finland, Kuopio, Finland Correspondence should be addressed to J J Palvimo: [email protected] Abstract Steroid receptors (SRs) constitute an important class of signal-dependent transcription Key Words factors (TFs). They regulate a variety of key biological processes and are crucial drug f androgen receptor targets in many disease states. In particular, estrogen (ER) and androgen receptors (AR) f estrogen receptor drive the development and progression of breast and prostate cancer, respectively. f glucocorticoid receptor Thus, they represent the main specific drug targets in these diseases. Recent evidence f progesterone receptor has suggested that the crosstalk between signal-dependent TFs is an important step f breast cancer in the reprogramming of chromatin sites; a signal-activated TF can expand or restrict f prostate cancer the chromatin binding of another TF. This crosstalk can rewire gene programs and thus f chromatin alter biological processes and influence the progression of disease. Lately, it has been f crosstalk postulated that there may be an important crosstalk between the AR and the ER with other SRs. Especially, progesterone (PR) and glucocorticoid receptor (GR) can reprogram chromatin binding of ER and gene programs in breast cancer cells. Furthermore, GR can take the place of AR in antiandrogen-resistant prostate cancer cells. Here, we review the current knowledge of the crosstalk between SRs in breast and prostate cancers. -

Clinicopathological and Genomic Features in Patients with Head and Neck Neuroendocrine Carcinoma
www.nature.com/modpathol ARTICLE OPEN Clinicopathological and genomic features in patients with head and neck neuroendocrine carcinoma Akihiro Ohmoto1, Yukiko Sato 2, Reimi Asaka2,3, Naoki Fukuda1, Xiaofei Wang1, Tetsuya Urasaki1, Naomi Hayashi1, Yasuyoshi Sato1, 1 1 1 1 4 5 2,3,6 Kenji Nakano , Mayu Yunokawa , Makiko✉ Ono , Junichi Tomomatsu , Takashi Toshiyasu , Hiroki Mitani , Kengo Takeuchi , Seiichi Mori7 and Shunji Takahashi1 © The Author(s) 2021 Neuroendocrine carcinoma (NEC) of the head and neck is a rare type of malignancy, accounting for only 0.3% of all head and neck cancers, and its clinicopathological and genomic features have not been fully characterized. We conducted a retrospective analysis of 27 patients with poorly differentiated NEC of the head and neck seen at our institution over a period of 15 years. Patient characteristics, adopted therapies, and clinical outcomes were reviewed based on the medical records. Pathological analysis and targeted sequencing of 523 cancer-related genes were performed using evaluable biopsied/resected specimens based on the clinical data. The most common tumor locations were the paranasal sinus (33%) and the oropharynx (19%). Eighty-one percent of the patients had locally advanced disease. The 3-year overall survival rates in all patients and in the 17 patients with locally advanced disease who received multimodal curative treatments were 39% and 53%, respectively. Histologically, large cell neuroendocrine carcinoma was the predominant subtype (58% of evaluable cases), and the Ki-67 labeling index ranged from 59 to 99% (median: 85%). Next-generation sequencing in 14 patients identified pathogenic/likely pathogenic variants in TP53, RB1, PIK3CA-related genes (PREX2, PIK3CA, and PTEN), NOTCH1, and SMARCA4 in six (43%), three (21%), two (14%), two (14%), and one (7%) patients, respectively. -

Pan-Cancer Transcriptome Analysis Reveals a Gene Expression
Modern Pathology (2016) 29, 546–556 546 © 2016 USCAP, Inc All rights reserved 0893-3952/16 $32.00 Pan-cancer transcriptome analysis reveals a gene expression signature for the identification of tumor tissue origin Qinghua Xu1,6, Jinying Chen1,6, Shujuan Ni2,3,4,6, Cong Tan2,3,4,6, Midie Xu2,3,4, Lei Dong2,3,4, Lin Yuan5, Qifeng Wang2,3,4 and Xiang Du2,3,4 1Canhelp Genomics, Hangzhou, Zhejiang, China; 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China; 3Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, China; 4Institute of Pathology, Fudan University, Shanghai, China and 5Pathology Center, Shanghai General Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China Carcinoma of unknown primary, wherein metastatic disease is present without an identifiable primary site, accounts for ~ 3–5% of all cancer diagnoses. Despite the development of multiple diagnostic workups, the success rate of primary site identification remains low. Determining the origin of tumor tissue is, thus, an important clinical application of molecular diagnostics. Previous studies have paved the way for gene expression-based tumor type classification. In this study, we have established a comprehensive database integrating microarray- and sequencing-based gene expression profiles of 16 674 tumor samples covering 22 common human tumor types. From this pan-cancer transcriptome database, we identified a 154-gene expression signature that discriminated the origin of tumor tissue with an overall leave-one-out cross-validation accuracy of 96.5%. The 154-gene expression signature was first validated on an independent test set consisting of 9626 primary tumors, of which 97.1% of cases were correctly classified. -

Regulation of the Oxidative Stress Response by Arid1a
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2015 REGULATION OF THE OXIDATIVE STRESS RESPONSE BY ARID1A Suet Yan Kwan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Oncology Commons Recommended Citation Kwan, Suet Yan, "REGULATION OF THE OXIDATIVE STRESS RESPONSE BY ARID1A" (2015). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 608. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/608 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. REGULATION OF THE OXIDATIVE STRESS RESPONSE BY ARID1A By Suet Yan Kwan, BSc APPROVED: ______________________________ Kwong-Kwok Wong, Ph.D. Advisory Professor ______________________________ Russell Broaddus, M.D., Ph.D. ______________________________ Joya Chandra, Ph.D. -

ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor?
Published OnlineFirst December 3, 2012; DOI: 10.1158/2159-8290.CD-12-0361 MINI REVIEW ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Jennifer N. Wu and Charles W.M. Roberts ABSTRACT Although disordered chromatin organization has long been recognized as a fea- ture of cancer, the molecular underpinnings of chromatin structure, epigenetic regulation, and their relationships to transcription are only beginning to be understood. Cancer genome sequencing studies have revealed a novel theme: frequent mutation of epigenetic regulators. Among these, the ARID1A/BAF250A subunit of the SWI/SNF (BRG1-associated factors) chromatin remod- eling complex has emerged as recurrently mutated in a broad array of tumor types. We review the genomic and functional data supporting classifi cation of ARID1A as a tumor suppressor. Signifi cance: Mutations in chromatin remodeling complex genes are increasingly recognized in many cancer types. However, the mechanisms by which chromatin remodeling complexes contribute to gene expression and the cancer phenotype are poorly understood. Understanding how mutation of chroma- tin remodelers facilitates transformation may offer the potential for development and implementation of novel therapies for cancer. Cancer Discov; 3(1); 35–43. ©2012 AACR. INTRODUCTION risk of developing malignant rhabdoid tumors at an especially young age. Epigenetic regulators impose upon the genetic code a Subsequent to these discoveries, implementation of next- chromatin structure characterized by chromatin accessibil- generation sequencing technologies have revealed frequent ity, nucleosome position, and histone modifi cations. These and recurrent mutations in a wide variety of epigenetic features of chromatin structure modulate gene expres- modulators, including mediators of DNA methylation (e.g., sion, thereby affecting both the identity and function of DNMT3a) and covalent modifi ers of histones (e.g., MLL, MLL3, cells. -

ARID1A Gene AT-Rich Interaction Domain 1A
ARID1A gene AT-rich interaction domain 1A Normal Function The ARID1A gene provides instructions for making a protein that forms one piece ( subunit) of several different SWI/SNF protein complexes. SWI/SNF complexes regulate gene activity (expression) by a process known as chromatin remodeling. Chromatin is the network of DNA and protein that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. Chromatin remodeling is one way gene expression is regulated during development; when DNA is tightly packed, gene expression is lower than when DNA is loosely packed. Through their ability to regulate gene activity, SWI/SNF complexes are involved in many processes, including repairing damaged DNA; copying (replicating) DNA; and controlling the growth, division, and maturation (differentiation) of cells. The ARID1A protein and other SWI/SNF subunits are thought to act as tumor suppressors, which keep cells from growing and dividing too rapidly or in an uncontrolled way. The ARID1A subunit is able to attach (bind) to DNA and is thought to help target SWI/ SNF complexes to the chromatin location that needs to be remodeled. Health Conditions Related to Genetic Changes Coffin-Siris syndrome More than 30 variants (also known as mutations) in the ARID1A gene can cause Coffin- Siris syndrome. This condition is characterized by delayed development, abnormalities of the fifth (pinky) fingers or toes, and characteristic facial features that are described as coarse. The ARID1A gene variants involved in Coffin-Siris syndrome lead to an abnormally short, nonfunctional protein. As a result, affected individuals have half the normal amount of functioning ARID1A protein. -
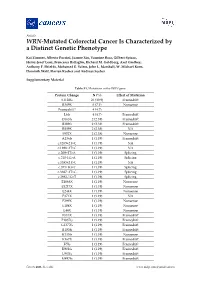
WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype
Article WRN-Mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype Kai Zimmer, Alberto Puccini, Joanne Xiu, Yasmine Baca, Gilbert Spizzo, Heinz-Josef Lenz, Francesca Battaglin, Richard M. Goldberg, Axel Grothey, Anthony F. Shields, Mohamed E. Salem, John L. Marshall, W. Michael Korn, Dominik Wolf, Florian Kocher and Andreas Seeber Supplementary Material Table S1. Mutations in the WRN gene. Protein Change N (%) Effect of Mutation S1128fs 26 (30.9) Frameshift R369X 6 (7.1) Nonsense Frameshift* 4 (4.7) L6fs 4 (4.7) Frameshift D163fs 2 (2.38) Frameshift R389fs 2 (2.38) Frameshift R889X 2 (2.38) NA S952X 2 (2.38) Nonsense A136fs 1 (1.19) Frameshift c.1269+2T>C 1 (1.19) NA c.1898+2T>G 1 (1.19) NA c.209+2T>A 1 (1.19) Splicing c.210-1G>A 1 (1.19) Splicing c.3383+2T>C 1 (1.19) NA c.355+1G>T 1 (1.19) Splicing c.3687+2T>G 1 (1.19) Splicing c.3983-1G>T 1 (1.19) Splicing E1068X 1 (1.19) Nonsense E1217X 1 (1.19) Nonsense E244X 1 (1.19) Nonsense E371X 1 (1.19) NA E399X 1 (1.19) Nonsense E488X 1 (1.19) Nonsense E48X 1 (1.19) Nonsense E513X 1 (1.19) Frameshift F1037fs 1 (1.19) Frameshift G1377fs 1 (1.19) Frameshift I1183fs 1 (1.19) Frameshift K135fs 1 (1.19) Nonsense K167X 1 (1.19) Frameshift K5fs 1 (1.19) Frameshift K901fs 1 (1.19) Frameshift L967fs 1 (1.19) Frameshift M497fs 1 (1.19) Frameshift Cancers 2020, 12, x; doi: www.mdpi.com/journal/cancers Cancers 2020, 12 S2 of 13 N166fs 1 (1.19) Frameshift Q11fs 1 (1.19) Frameshift R1305X 1 (1.19) Nonsense R279fs 1 (1.19) Frameshift R565X 1 (1.19) Nonsene R741X 1 (1.19) Nonsense R987X 1 (1.19) NA T1011fs 1 (1.19) Frameshift W1014X 1 (1.19) Nonsense Y57X 1 (1.19) Nonsense Grand Total 84 *4 Frameshift mutations could not be described more precisely.