A Review of Acetaminophen Poisoning
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management of Poisoning
MOH CLINICAL PRACTICE GUIDELINES December/2011 Management of Poisoning Health Ministry of Sciences Chapter of Emergency College of College of Family Manpower Authority Physicians Physicians, Physicians Academy of Medicine, Singapore Singapore Singapore Singapore Medical Pharmaceutical Society Society for Emergency Toxicology Singapore Paediatric Association of Singapore Medicine in Singapore Society (Singapore) Society Executive summary of recommendations Details of recommendations can be found in the main text at the pages indicated. Principles of management of acute poisoning – resuscitating the poisoned patient GPP In a critically poisoned patient, measures beyond standard resuscitative protocol like those listed above need to be implemented and a specialist experienced in poisoning management should be consulted (pg 55). GPP D Prolonged resuscitation should be attempted in drug-induced cardiac arrest (pg 55). Grade D, Level 3 1 C Titrated doses of naloxone, together with bag-valve-mask ventilation, should be administered for suspected opioid-induced coma, prior to intubation for respiratory insuffi ciency (pg 56). Grade C, Level 2+ D In bradycardia due to calcium channel or beta-blocker toxicity that is refractory to conventional vasopressor therapy, intravenous calcium, glucagon or insulin should be used (pg 57). Grade D, Level 3 B Patients with actual or potential life threatening cardiac arrhythmia, hyperkalaemia or rapidly progressive toxicity from digoxin poisoning should be treated with digoxin-specifi c antibodies (pg 57). Grade B, Level 2++ B Titrated doses of benzodiazepine should be given in hyperadrenergic- induced tachycardia states resulting from poisoning (pg 57). Grade B, Level 1+ D Non-selective beta-blockers, like propranolol, should be avoided in stimulant toxicity as unopposed alpha agonism may worsen accompanying hypertension (pg 57). -

Poisoning in Children
ARTICLE IN PRESS Current Paediatrics (2005) 15, 563–568 www.elsevier.com/locate/cupe Poisoning in children Fiona JepsenÃ, Mary Ryan Emergency Medicine, Royal Liverpool Children’s NHS Trust, Alder Hey, Liverpool L12 2AP, UK KEYWORDS Summary Poisoning accounts for about 7% of all accidents in children under 5 Poisoning; years and is implicated in about 2% of all childhood deaths in the developed world, Child; and over 5% in the developing world (National Poisons Information Service). In Accidents; considering this topic, however, it is important to differentiate accidental overdose Home (common in the younger age groups) and deliberate overdose (more common in young adults). Although initial assessment and treatment of these groups may not differ significantly, the social issues and ongoing follow-up of these children will be totally different and the treating physician must remain aware of this difference. The initial identification and treatment of these children remains the mainstay of management, and many ingested substances do not have a specific antidote. Supportive treatment must be planned and the potential for delayed or long-term effects noted. The specific presentation and treatment of some of the commonly ingested substances will be addressed in this article, and guidance given on when to contact expert help. & 2005 Elsevier Ltd. All rights reserved. Introduction such as bleaches, detergents and turpentine sub- stitutes. More than 100 000 individuals are admitted to Toxic compounds may be ingested or inhaled hospital in England and Wales annually due to either accidentally or deliberately. Accidental poisoning, accounting for 10% of all acute admis- poisoning can occur at any age, but is much more 1 sions.1 However, the true incidence of acute common in children. -

Acute Poisoning: Understanding 90% of Cases in a Nutshell S L Greene, P I Dargan, a L Jones
204 REVIEW Postgrad Med J: first published as 10.1136/pgmj.2004.027813 on 5 April 2005. Downloaded from Acute poisoning: understanding 90% of cases in a nutshell S L Greene, P I Dargan, A L Jones ............................................................................................................................... Postgrad Med J 2005;81:204–216. doi: 10.1136/pgmj.2004.024794 The acutely poisoned patient remains a common problem Paracetamol remains the most common drug taken in overdose in the UK (50% of intentional facing doctors working in acute medicine in the United self poisoning presentations).19 Non-steroidal Kingdom and worldwide. This review examines the initial anti-inflammatory drugs (NSAIDs), benzodiaze- management of the acutely poisoned patient. Aspects of pines/zopiclone, aspirin, compound analgesics, drugs of misuse including opioids, tricyclic general management are reviewed including immediate antidepressants (TCAs), and selective serotonin interventions, investigations, gastrointestinal reuptake inhibitors (SSRIs) comprise most of the decontamination techniques, use of antidotes, methods to remaining 50% (box 1). Reductions in the price of drugs of misuse have led to increased cocaine, increase poison elimination, and psychological MDMA (ecstasy), and c-hydroxybutyrate (GHB) assessment. More common and serious poisonings caused toxicity related ED attendances.10 Clinicians by paracetamol, salicylates, opioids, tricyclic should also be aware that severe toxicity can result from exposure to non-licensed pharmaco- -

Cheminformatics Tools for Enabling Metabolomics by Yannick Djoumbou Feunang
Cheminformatics Tools for Enabling Metabolomics by Yannick Djoumbou Feunang A thesis submitted in partial fulfillment of requirements for the degree of Doctor of Philosophy in Microbiology and Biotechnology Department of Biological Sciences University of Alberta ©Yannick Djoumbou Feunang, 2017 ii Abstract Metabolites are small molecules (<1500 Da) that are used in or produced during chemical reactions in cells, tissues, or organs. Upon absorption or biosynthesis in humans (or other organisms), they can either be excreted back into the environment in their original form, or as a pool of degradation products. The outcome and effects of such interactions is function of many variables, including the structure of the starting metabolite, and the genetic disposition of the host organism. For this reasons, it is usually very difficult to identify the transformation products as well as their long-term effect in humans and the environment. This can be explained by many factors: (1) the relevant knowledge and data are for the most part unavailable in a publicly available electronic format; (2) when available, they are often represented using formats, vocabularies, or schemes that vary from one source (or repository) to another. Assuming these issues were solved, detecting patterns that link the metabolome to a specific phenotype (e.g. a disease state), would still require that the metabolites from a biological sample be identified and quantified, using metabolomic approaches. Unfortunately, the amount of compounds with publicly available experimental data (~20,000) is still very small, compared to the total number of expected compounds (up to a few million compounds). For all these reasons, the development of cheminformatics tools for data organization and mapping, as well as for the prediction of biotransformation and spectra, is more crucial than ever. -

Hospital Formulary (List of Drugs, Chemicals & Dressing Material)
Hospital Formulary (List of drugs, chemicals & dressing material) Department of Pharmacy Government Medical College Hospital Sector-32, Chandigarh Compiled by: Jayati Khurana (M.Pharmacy in Pharmaceutics) Department of Pharmacy - In The Service of Humanity Prof Ravi Gupta Head Pharmacy Ms. Neetu Verma Dispensary Superintendent Ms. Manjeet Kaur Pharmacist Ms. Kuldeep Kaur Pharmacist Ms. Neelam Pharmacist Ms. Parveen Lata Pharmacist Ms. Monika Verma Pharmacist Mr. Jatinder Singh Pharmacist Ms. Bharti Rawat Pharmacist Ms. Neetu Verma Pharmacist Mr. Satinder Parkash Pharmacist Mr. Ravinder Pharmacist Ms. Rachna Bisht Pharmacist Ms. Alka Sinhmar Pharmacist Ms. Nisha Rani Pharmacist Ms. Pooja Pharmacist Ms. Charu Pharmacist Ms. Jayati Khurana Pharmacist Ms. Monika Yadav Pharmacist Ms. Kala Wanti Senior Assistant Ms. Vandana Junior Assistant Mr. Chhinder Data Entry Operator INDEX S. No. Pharmacological Category Page number 1 Abortifacients/Uterine stimulants 1 2 Alkalizing agent 1 3 Alpha-1 Blocker 1 4 Aminoglycoside antibiotics (Bactericidal) 1 5 Analgesic-Anti-inflammatory drugs 2-3 6 Antacids 3 7 Anti-acne drugs 4 8 Anti-allergic drugs 4 9 Anti-amoebic drugs 4 10 Antianginal drugs 4-5 11 Anti-anxiety drugs 5 12 Antiarrhythmic drugs 6 13 Antiarrhythmics-Local Anesthetics 6 14 Antiasthmatic drugs 6-8 15 Anticancer drugs 8-10 16 Anticholinergic drugs 10 17 Anti-coagulant 10-11 18 Antidepressants 11 19 Antidiarrhoeal drugs 12 20 Anti-Diuretic Hormone 12 21 Antidotes 12-13 22 Anti-Emetic drugs 13-14 23 Anti-Epileptics 14-15 24 Antifungal drugs 15-16 25 Anthelminthic drugs 16 26 Antihistamines 17 27 Anti-hyperglycemics 17 28 Antihypertensive drugs 18 29 Antiinflammatory-local anesthetics 18 30 Anti-leishmaniasis (Kala-azar) 18 31 Anti-leprotics 19 32 Anti-malarial drugs 19-20 33 Antimaniac drugs 20 34 Antiparkison drugs 21 35 Anti-peptic ulcers 21-22 36 Antiplatelets 22 37 Anti-psoriatics 22 38 Anti-psychotics 22-23 39 Anti-pyretics 23 40 Anti-scabies/Anti-lice 23 41 Antiseptic-Disinfectants 23-24 42 Antispasmodics 24-25 43 Anti-T.B. -

Pattern of Paracetamol Poisoning: Influence on Outcome and Complications
toxics Article Pattern of Paracetamol Poisoning: Influence on Outcome and Complications Diego Castanares-Zapatero 1, Valérie Dinant 1, Ilaria Ruggiano 1, Harold Willem 1, Pierre-François Laterre 1 and Philippe Hantson 1,2,* 1 Department of Intensive Care, Cliniques St-Luc, Université catholique de Louvain, 1200 Brussels, Belgium; [email protected] (D.C.-Z.); [email protected] (V.D.); [email protected] (I.R.); [email protected] (H.W.); [email protected] (P.-F.L.) 2 Louvain Centre for Toxicology and Applied Pharmacology, 1200 Brussels, Belgium * Correspondence: [email protected]; Tel.: +00-327-642-755 Received: 5 September 2018; Accepted: 28 September 2018; Published: 29 September 2018 Abstract: Acute paracetamol poisoning due to a single overdose may be effectively treated by the early administration of N-acetylcysteine (NAC) as an antidote. The prognosis may be different in the case of intoxication due to multiple ingestions or when the antidote is started with delay. The aim of this work was to investigate the outcome of paracetamol poisoning according to the pattern of ingestion and determine the factors associated with the outcome. We performed a retrospective analysis over the period 2007–2017 of the patients who were referred to a tertiary hospital for paracetamol-related hepatotoxicity. Inclusion criteria were: accidental or voluntary ingestion of paracetamol, delay for NAC therapy of 12 h or more, liver enzymes (ALT) >1000 IU/L on admission. Ninety patients were considered. Poisoned patients following multiple ingestion were significantly older (45 ± 12 vs. 33 ± 14) (p = 0.001), with a higher incidence of liver steatosis (p = 0.016) or chronic ethanol abuse (p = 0.04). -

Question of the Day Archives: Monday, December 5, 2016 Question: Calcium Oxalate Is a Widespread Toxin Found in Many Species of Plants
Question Of the Day Archives: Monday, December 5, 2016 Question: Calcium oxalate is a widespread toxin found in many species of plants. What is the needle shaped crystal containing calcium oxalate called and what is the compilation of these structures known as? Answer: The needle shaped plant-based crystals containing calcium oxalate are known as raphides. A compilation of raphides forms the structure known as an idioblast. (Lim CS et al. Atlas of select poisonous plants and mushrooms. 2016 Disease-a-Month 62(3):37-66) Friday, December 2, 2016 Question: Which oral chelating agent has been reported to cause transient increases in plasma ALT activity in some patients as well as rare instances of mucocutaneous skin reactions? Answer: Orally administered dimercaptosuccinic acid (DMSA) has been reported to cause transient increases in ALT activity as well as rare instances of mucocutaneous skin reactions. (Bradberry S et al. Use of oral dimercaptosuccinic acid (succimer) in adult patients with inorganic lead poisoning. 2009 Q J Med 102:721-732) Thursday, December 1, 2016 Question: What is Clioquinol and why was it withdrawn from the market during the 1970s? Answer: According to the cited reference, “Between the 1950s and 1970s Clioquinol was used to treat and prevent intestinal parasitic disease [intestinal amebiasis].” “In the early 1970s Clioquinol was withdrawn from the market as an oral agent due to an association with sub-acute myelo-optic neuropathy (SMON) in Japanese patients. SMON is a syndrome that involves sensory and motor disturbances in the lower limbs as well as visual changes that are due to symmetrical demyelination of the lateral and posterior funiculi of the spinal cord, optic nerve, and peripheral nerves. -

Publications for Nicholas Buckley 2021 2020
Publications for Nicholas Buckley 2021 href="http://dx.doi.org/10.1002/prp2.695">[More Brett, J., Pearson, S., Daniels, B., Wylie, C., Buckley, N. Information]</a> (2021). A cross sectional study of psychotropic medicine use in Crouse, J., Morley, K., Buckley, N., Dawson, A., Seth, D., Australia in 2018: A focus on polypharmacy. British Journal of Monds, L., Tickell (nee Kennedy), A., Kay-Lambkin, F., Clinical Pharmacology, 87(3), 1369-1377. <a Chitty, K. (2021). Online interventions for people hospitalized href="http://dx.doi.org/10.1111/bcp.14527">[More for deliberate self-harm and problematic alcohol use: Lessons Information]</a> learned from the iiAIM trial. Bulletin of the Menninger Clinic, Chiew, A., Buckley, N. (2021). Acetaminophen Poisoning. 85(2), 123-142. <a Critical Care Clinics, 37(3), 543-561. <a href="http://dx.doi.org/10.1521/bumc.2021.85.2.123">[More href="http://dx.doi.org/10.1016/j.ccc.2021.03.005">[More Information]</a> Information]</a> Perananthan, V., Buckley, N. (2021). Opioids and Hurzeler, T., Buckley, N., Noghrehchi, F., Malouf, P., Page, A., antidepressants: Which combinations to avoid. Australian Schumann, J., Chitty, K. (2021). Alcohol-related suicide across Prescriber, 44(2), 41-44. <a Australia: a geospatial analysis. Australian and New Zealand href="http://dx.doi.org/10.18773/austprescr.2021.004">[More Journal of Public Health, 45(4), 394-399. <a Information]</a> href="http://dx.doi.org/10.1111/1753-6405.13122">[More Ly, J., Brown, J., Buckley, N., Cairns, R. (2021). Paediatric Information]</a> poisoning exposures in schools: Reports to Australia's largest Huang, J., Buckley, N., Isoardi, K., Chiew, A., Isbister, G., poisons centre. -

Exposure to Acetaminophen and All Its Metabolites Upon 10, 15, and 20Mg/Kg Intravenous Acetaminophen in Very-Preterm Infants
Articles | Clinical Investigation nature publishing group Exposure to acetaminophen and all its metabolites upon 10, 15, and 20mg/kg intravenous acetaminophen in very-preterm infants Robert B. Flint1,2,3,11, Daniella W. Roofthooft1,11,AnnevanRongen4,5, Richard A. van Lingen6, Johannes N. van den Anker7,8,9, Monique van Dijk1,7, Karel Allegaert1,7,10, Dick Tibboel7, Catherijne A.J. Knibbe4,5 and Sinno H.P. Simons1 BACKGROUND: Exposure to acetaminophen and its meta- acetaminophen as an opioid-sparing therapy in adults and bolites in very-preterm infants is partly unknown. We children has now been introduced in neonatal intensive care investigated the exposure to acetaminophen and its meta- units across the globe (4). However, only very limited data of bolites upon 10, 15, or 20 mg/kg intravenous acetaminophen its use are available in the most preterm infants (5,6). in preterm infants. Acetaminophen (N-acetyl-p-amino-phenol) is extensively METHODS: In a randomized trial, 59 preterm infants metabolized in the liver. The main pathways involved are (24–32 weeks’ gestational age, postnatal age o1 week) glucuronidation and sulfation, which in adults account for received 10, 15, or 20 mg/kg acetaminophen intravenously. ~ 55 and 30% of acetaminophen metabolism, respectively Plasma concentrations of acetaminophen and its metabolites (7–9) (Figure 1). Only 2–5% is excreted unchanged in the (glucuronide, sulfate, cysteine, mercapturate, and glutathione) urine. Approximately 5–10% of acetaminophen is metabo- were determined in 293 blood samples. Area under the lized by cytochrome P450 (CYP), primarily by the CYP2E1 – plasma concentration time curves (AUC0–500 min) was related enzyme (10–12), to the toxic metabolite N-acetyl-p-benzo- to dose and gestational age. -
Random2 Mcqs
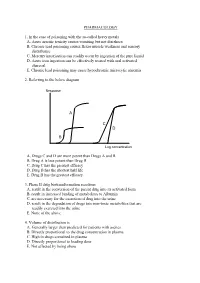
PHARMACOLOGY 1. In the case of poisoning with the so-called heavy metals A. Acute arsenic toxicity causes vomiting but not diarrhoea B. Chronic lead poisoning causes flexor muscle weakness and sensory disturbance C. Mercury intoxication can readily occur by ingestion of the pure liquid D. Acute iron ingestion can be effectively treated with oral activated charcoal E. Chronic lead poisoning may cause hypochromic microcytic anaemia 2. Referring to the below diagram Response A C D B Log concentration A. Drugs C and D are more potent than Drugs A and B B. Drug A is less potent than Drug B C. Drug C has the greatest efficacy D. Drug B has the shortest half life E. Drug B has the greatest efficacy 3. Phase II drug biotransformation reactions A. result in the conversion of the parent drug into its activated form B. result in increased binding of metabolites to Albumin C. are necessary for the excretion of drug into the urine D. result in the degradation of drugs into non-toxic metabolites that are readily excreted into the urine E. None of the above 4. Volume of distribution is A. Generally larger than predicted for patients with ascites B. Directly proportional to the drug concentration in plasma C. High in drugs contained in plasma D. Directly proportional to loading dose E. Not affected by being obese 5. Suxamethonium A. Has no effect on heart rate B. May cause decreased intraocular pressure C. Would be the muscle relaxant of choice in a patient severely burned two weeks prior D. Has a short duration of action due to rapid hydrolysis by acetyl cholinesterase E. -

Sign up to Receive ATOTW Weekly – Email [email protected]
Sign up to receive ATOTW weekly – email [email protected] ACCIDENTAL POISONING IN CHILDREN ANAESTHESIA TUTORIAL OF THE WEEK 95 9TH JUNE 2008 Dr Susara Ribbens Specialist Registrar North Central London School of Anaesthetics [email protected] Introduction Poisoning is a significant global public health problem. According to the World Health Organisation data, an estimated 350 000 people died from unintentional poisoning in 2002. Accidental poisoning occurs most often in the age group 1-5 years although less than one per cent of poisoning in children is serious. More than 94% of fatal poisonings occur in low- and middle-income countries. It remains a challenge to identify those at risk at an early stage. Education of the general public and also the provision of child-proof containers for household chemicals and medicines play a key role in prevention. (Toxic plants, insect and snake bites are not discussed in this tutorial, but see www.rch.org.au/poisons for more information) Questions True or false: 1. Emesis or nasogastric aspiration are indicated after sodium hydroxide ingestion. 2. Regarding paraffin ingestion – abnormalities on the chest film may be seen in the absence of symptoms. 3. A fever after paraffin ingestion is always due to a secondary infection. 4. Ingestion of small amounts of paraquat is usually harmless. 5. High FiO2 should be avoided in paraquat ingestion to prevent pulmonary fibrosis. 6. Organophosphate forms an irreversible complex with cholinesterase. 7. Carbon monoxide is irritant with a distinctive odour. 8. The SpO2 reading in carbon monoxide poisoning is usually less than 85%. 9. A plasma paracetamol level should be taken within 2 hours of ingestion. -

Update of Toxicology and Toxinology Guidelines
What’s new in Toxicology and Toxinology Guidelines? Dr Venita Munir Editor [email protected] Therapeutic Guidelines Ltd. ▪ Not-for-profit, financially self-supporting, intellectually independent ▪ Improve patient outcomes by promoting the quality use of medicines ▪ Create and publish point-of-care guidelines ▪ Products: ▪ eTG complete (since 2002) ▪ eTG app (since 2016) ▪ books phased out (2019) Toxicology and Toxinology 3 ▪ Not updated since 2012 ▪ Expert group assembled in late 2017 Toxicology and Toxinology Expert Group Clinical Toxicologists / Emergency Physicians: Prof. Nick Buckley (NSW) Chair Prof. Geoff Isbister (NSW) Dr Sam Alfred (SA) Dr Katherine Isoardi (QLD) Dr Angela Chiew (NSW) Dr Zeff Koutsogiannis (VIC) Prof. Andrew Dawson (NSW) Dr Jess Soderstrom (WA) Prof. Andis Graudins (VIC) Specialists in Poisons Information (SPIs): Mr Kasra Ahmadi (WA) Ms Elizabeth Nguyen (NSW) Dr Dawson Macleod (VIC) Ms Carol Wylie (QLD) General Practitioner: Dr Marion Christie (VIC) What’s new in Toxicology and Toxinology? • 54 Existing monographs and 38 New monographs • Many topics split & linked to multiple specific topics • More structure: • Management overview • Risk assessment • Treatment • Observation & Disposition Overview of changes • Covers more rare but highly toxic agents • More detail overall; eg: • Toxic doses and concentrations • Specific decontamination advice • Links to General approach & Toxidromes • Paediatric doses and maximums • Less emphasis on antidotes • Specific advice on duration of observation Existing monographs updated