ORGANIC CHEMISTRY Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding
Structure and Bonding 174 Series Editor: D.M.P. Mingos Stefanie Dehnen Editor Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding Series Editor: D.M.P. Mingos, Oxford, United Kingdom Editorial Board: X. Duan, Beijing, China L.H. Gade, Heidelberg, Germany Y. Lu, Urbana, IL, USA F. Neese, Mulheim€ an der Ruhr, Germany J.P. Pariente, Madrid, Spain S. Schneider, Gottingen,€ Germany D. Stalke, Go¨ttingen, Germany Aims and Scope Structure and Bonding is a publication which uniquely bridges the journal and book format. Organized into topical volumes, the series publishes in depth and critical reviews on all topics concerning structure and bonding. With over 50 years of history, the series has developed from covering theoretical methods for simple molecules to more complex systems. Topics addressed in the series now include the design and engineering of molecular solids such as molecular machines, surfaces, two dimensional materials, metal clusters and supramolecular species based either on complementary hydrogen bonding networks or metal coordination centers in metal-organic framework mate- rials (MOFs). Also of interest is the study of reaction coordinates of organometallic transformations and catalytic processes, and the electronic properties of metal ions involved in important biochemical enzymatic reactions. Volumes on physical and spectroscopic techniques used to provide insights into structural and bonding problems, as well as experimental studies associated with the development of bonding models, reactivity pathways and rates of chemical processes are also relevant for the series. Structure and Bonding is able to contribute to the challenges of communicating the enormous amount of data now produced in contemporary research by producing volumes which summarize important developments in selected areas of current interest and provide the conceptual framework necessary to use and interpret mega- databases. -

(T = C/Si/Ge): the Uniqueness of Carbon Bonds in Tetrel Bonds
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 13 September 2018 doi:10.20944/preprints201809.0228.v1 Inter/intramolecular Bonds in TH5+ (T = C/Si/Ge): The Uniqueness of Carbon bonds in Tetrel Bonds Sharon Priya Gnanasekar and Elangannan Arunan* Department of Inorganic and Physical Chemistry Indian Institute of Science, Bangalore. 560012 INDIA * Email: [email protected] Abstract Atoms in Molecules (AIM), Natural Bond Orbital (NBO), and normal coordinate analysis have been carried out at the global minimum structures of TH5+ (T = C/Si/Ge). All these analyses lead to a consistent structure for these three protonated TH4 molecules. The CH5+ has a structure with three short and two long C-H covalent bonds and no H-H bond. Hence, the popular characterization of protonated methane as a weakly bound CH3+ and H2 is inconsistent with these results. However, SiH5+ and GeH5+ are both indeed a complex formed between TH3+ and H2 stabilized by a tetrel bond, with the H2 being the tetrel bond acceptor. The three-center-two-electron bond (3c-2e) in CH5+ has an open structure, which can be characterized as a V-type 3c-2e bond and that found in SiH5+ and GeH5+ is a T-type 3c-2e bond. This difference could be understood based on the typical C-H, Si-H, Ge-H and H-H bond energies. Moreover, this structural difference observed in TH5+ can explain the trend in proton affinity of TH4. Carbon is selective in forming a ‘tetrel bond’ and when it does, it might be worthwhile to highlight it as a ‘carbon bond’. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

Proton-Coupled Reduction of N2 Facilitated by Molecular Fe Complexes
Proton-Coupled Reduction of N2 Facilitated by Molecular Fe Complexes Thesis by Jonathan Rittle In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2016 (Defended November 9, 2015) ii 2016 Jonathan Rittle All Rights Reserved iii ACKNOWLEDGEMENTS My epochal experience in graduate school has been marked by hard work, intellectual growth, and no shortage of deadlines. These facts leave little opportunity for an acknowledgement of those whose efforts were indispensable to my progress and perseverance, and are therefore disclosed here. First and foremost, I would like to thank my advisor, Jonas Peters, for his tireless efforts in molding me into the scientist that I am today. His scientific rigor has constantly challenged me to strive for excellence and I am grateful for the knowledge and guidance he has provided. Jonas has given me the freedom to pursue scientific research of my choosing and shown a remarkable degree of patience while dealing with my belligerent tendencies. Perhaps equally important, Jonas has built a research group that is perpetually filled with the best students and postdoctoral scholars. Their contributions to my graduate experience are immeasurable and I wish them all the best of luck in their future activities. In particular, John Anderson, Dan Suess, and Ayumi Takaoka were senior graduate students who took me under their wings when I joined the lab as a naïve first-year graduate student. I am grateful for the time and effort that they collectively spent in teaching me the art of chemical synthesis and for our unforgettable experiences outside of lab. -

Is There an Acid Strong Enough to Dissolve Glass? – Superacids
ARTICLE Is there an acid strong enough to dissolve glass? – Superacids For anybody who watched cartoons growing up, the word unit is based on how acids behave in water, however as acid probably springs to mind images of gaping holes being very strong acids react extremely violently in water this burnt into the floor by a spill, and liquid that would dissolve scale cannot be used for the pure ‘common’ acids (nitric, anything you drop into it. The reality of the acids you hydrochloric and sulphuric) or anything stronger than them. encounter in schools, and most undergrad university Instead, a different unit, the Hammett acidity function (H0), courses is somewhat underwhelming – sure they will react is often preferred when discussing superacids. with chemicals, but, if handled safely, where’s the drama? A superacid can be defined as any compound with an People don’t realise that these extraordinarily strong acids acidity greater than 100% pure sulphuric acid, which has a do exist, they’re just rarely seen outside of research labs Hammett acidity function (H0) of −12 [1]. Modern definitions due to their extreme potency. These acids are capable of define a superacid as a medium in which the chemical dissolving almost anything – wax, rocks, metals (even potential of protons is higher than it is in pure sulphuric acid platinum), and yes, even glass. [2]. Considering that pure sulphuric acid is highly corrosive, you can be certain that anything more acidic than that is What are Superacids? going to be powerful. What are superacids? Its all in the name – super acids are intensely strong acids. -
Bonding in Coordination Compounds
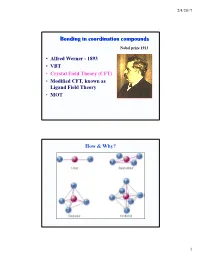
2/4/2017 Bonding in coordination compounds Nobel prize 1913 • Alfred Werner - 1893 • VBT • Crystal Field Theory (CFT) • Modified CFT, known as Ligand Field Theory • MOT How & Why? 1 2/4/2017 Valance Bond Theory Basic Principle A covalent bond forms when the orbtials of two atoms overlap and are occupied by a pair of electrons that have the highest probability of being located between the nuclei. Linus Carl Pauling (1901-1994) Nobel prizes: 1954, 1962 Valance Bond Model Ligand = Lewis base Metal = Lewis acid s, p and d orbitals give hybrid orbitals with specific geometries Number and type of M-L hybrid orbitals determines geometry of the complex Octahedral Complex 3+ e.g. [Cr(NH3)6] 2 2/4/2017 2- 2- Tetrahedral e.g. [Zn(OH)4] Square Planar e.g. [Ni(CN)4] Limitations of VB theory Cannot account for colour of complexes May predict magnetism wrongly Cannot account for spectrochemical series Crystal Field Theory 400 500 600 800 •The relationship between colors and complex metal ions 3 2/4/2017 Crystal Field Model A purely ionic model for transition metal complexes. Ligands are considered as point charge. Predicts the pattern of splitting of d-orbitals. Used to rationalize spectroscopic and magnetic properties. d-orbitals: look attentively along the axis Linear combination of 2 2 2 2 dz -dx and dz -dy 2 2 2 d2z -x -y 4 2/4/2017 Octahedral Field • We assume an octahedral array of negative charges placed around the metal ion (which is positive). • The ligand and orbitals lie on the same axes as negative charges. -

Chemical Science
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Highly selective acylation of polyamines and aminoglycosides by 5-acyl-5-phenyl-1,5-dihydro- Cite this: Chem. Sci.,2017,8,7152 4H-pyrazol-4-ones† Kostiantyn O. Marichev, Estevan C. Garcia, Kartick C. Bhowmick, Daniel J. Wherritt, Hadi Arman and Michael P. Doyle * 5-Acyl-5-phenyl-1,5-dihydro-4H-pyrazol-4-ones, accessible from arylpropargyl phenyldiazoacetates, are highly selective acyl transfer reagents for di- and polyamines, as well as aminoalcohols and aminothiols. As reagents with a carbon-based leaving group, they have been applied for benzoyl transfer with a broad selection of substrates containing aliphatic amino in combination with other competing nucleophilic Received 20th July 2017 functional groups. The substrate scope and levels of selectivity for direct benzoyl transfer exceed those Accepted 29th August 2017 of known benzoylating reagents. With exceptional selectivity for acylation between primary amines DOI: 10.1039/c7sc03184j bound to primary and secondary carbons, these new reagents have been used in direct site-selective Creative Commons Attribution 3.0 Unported Licence. rsc.li/chemical-science monobenzoylation of aminoglycoside antibiotics. Introduction 1-position of 1,2-diaminopropane,14d selective acylation of primary amines whose carbon attachment is primary, The formation of an amide bond by acyl transfer is a classic secondary or tertiary, has received scant attention.15 chemical reaction1 that has been extensively studied2 and widely We have recently prepared a novel heterocyclic compound that applied.3 Over the years numerous acyl transfer agents have been appeared to have the potential of being a selective benzoyl transfer investigated; their activities have been dependent on the leaving reagent. -

A TRANS INFLUENCE and Π-CONJUGATION EFFECTS on LIGAND SUBSTITUTION REACTIONS of Pt(II) COMPLEXES with TRIDENTATE PENDANT N/S-DONOR LIGANDS
Tanzania Journal of Science 44(2): 45-63, 2018 ISSN 0856-1761, e-ISSN 2507-7961 © College of Natural and Applied Sciences, University of Dar es Salaam, 2018 A TRANS INFLUENCE AND π-CONJUGATION EFFECTS ON LIGAND SUBSTITUTION REACTIONS OF Pt(II) COMPLEXES WITH TRIDENTATE PENDANT N/S-DONOR LIGANDS Grace A Kinunda Chemistry Department, University of Dar es Salaam, P. O. Box 35061, Dar es Salaam, Tanzania E-mail:[email protected]/[email protected] ABSTRACT The rate of displacement of the chloride ligands by three neutral nucleophiles (Nu) of different steric demands, namely thiourea (TU), N,N’-dimethylthiourea (DMTU) and N,N,N,’N- tetramethylthiourea (TMTU) in the complexes viz; [Pt(II)(bis(2-pyridylmethyl)amine)Cl]ClO4, (Pt1), [Pt(II){N-(2-pyridinylmethyl)-8-quinolinamine}Cl]Cl, (Pt2), [Pt(II)(bis(2- pyridylmethyl)sulfide)Cl]Cl, (Pt3) and [Pt(II){8-((2-pyridylmethyl)thiol)quinoline}Cl]Cl, (Pt4) was studied under pseudo first-order conditions as a function of concentration and temperature using a stopped-flow technique and UV-Visible spectrophotometry. The observed pseudo first- order rate constants for substitution reactions obeyed the simple rate law kobs k2Nu. The results have shown that the chloro ligand in Pt(N^S^N) complexes is more labile by two orders of magnitude than Pt(N^N^N) complexes due to the high trans labilizing effect brought by the S- donor atom. The quinoline based Pt(II) complexes (Pt2 and Pt4) have been found to be slow than their pyridine counterparts Pt1 and Pt3 due to poor π-acceptor ability of quinoline. -

1.5 Phosphines As Ligands in Transition Metal Complexes
_________________________________________________________________________Swansea University E-Theses The synthesis and characterisation of some novel compounds containing Pt-Se bonds. Webster, Christopher Alan How to cite: _________________________________________________________________________ Webster, Christopher Alan (2006) The synthesis and characterisation of some novel compounds containing Pt-Se bonds.. thesis, Swansea University. http://cronfa.swan.ac.uk/Record/cronfa43110 Use policy: _________________________________________________________________________ This item is brought to you by Swansea University. Any person downloading material is agreeing to abide by the terms of the repository licence: copies of full text items may be used or reproduced in any format or medium, without prior permission for personal research or study, educational or non-commercial purposes only. The copyright for any work remains with the original author unless otherwise specified. The full-text must not be sold in any format or medium without the formal permission of the copyright holder. Permission for multiple reproductions should be obtained from the original author. Authors are personally responsible for adhering to copyright and publisher restrictions when uploading content to the repository. Please link to the metadata record in the Swansea University repository, Cronfa (link given in the citation reference above.) http://www.swansea.ac.uk/library/researchsupport/ris-support/ The Synthesis and Characterisation of Some Novel Compounds Containing Pt-Se Bonds Christopher Alan Webster Department of Chemistry University of Wales Swansea November 2006 ProQuest Number: 10821502 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. -

Masterarbeit / Master's Thesis
MASTERARBEIT / MASTER’S THESIS Titel der Masterarbeit / Title of the Master‘s Thesis „Chemical Pathways in Protoplanetary Discs“ verfasst von / submitted by Robert Pohl BSc angestrebter akademischer Grad / in partial fulfilment of the requirements for the degree of Master of Science (MSc) Wien, 2016 / Vienna 2016 Studienkennzahl lt. Studienblatt / A 066 861 degree programme code as it appears on the student record sheet: Studienrichtung lt. Studienblatt / Astronomie UG 2002 degree programme as it appears on the student record sheet: Betreut von / Supervisor: Univ.-Prof. Dr. Manuel Güdel - 2 - And God said, “Let there be lights in the vault of the sky to separate the day from the night, and let them serve as signs to mark sacred times, and days and years, and let them be lights in the vault of the sky to give light on the earth.” And it was so. God made two great lights—the greater light to govern the day and the lesser light to govern the night. He also made the stars. God set them in the vault of the sky to give light on the earth, to govern the day and the night, and to separate light from darkness. And God saw that it was good. And there was evening, and there was morning—the fourth day. Genesis 1.14-19 This image shows IRAS 04302+2247; the edge-on disk of dust and gas has a diameter of ~ 800 AU and a mass comparable to the Solar Nebula, which gave birth to Sun’s planetary system. Dark clouds and bright wisps above and below the disk suggest that it is still building up from infalling dust and gas. -

8. Chemistry of the Main Group Elements Unusual Bonding
8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry unusual bonding , structure & reactivity 8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry very powerful reducing agent te 2+ a − in d r o ? o ! c n - o b six r a c 2− Na2 [Ne]3s2 {Ba-cryptand} + disodide 2− M.Y. Redko et al. JACS 2003 gold(I) methanium also, in NH3(l) H. Schmidbaur et al. Na + (NH ) e − Chem. Ber. 1992 3 x 1 8. Chemistry of the Main Group Elements A Snapshot on Main Group Chemistry very powerful reducing agent 2+ − 2− Na2 [Ne]3s2 {Ba-cryptand} + disodide 2− M.Y. Redko et al. JACS 2003 gold(I) methanium also, in NH3(l) H. Schmidbaur & F. Gabbai Na + (NH ) e − Chem. Ber. 1997 3 x 8. Chemistry of the Main Group Elements here we go again… table salt #1 …well, not in my book! Check out Nitrogenase or Cytochrome C-Oxidase…or Hemoglobin… 2 8. Chemistry of the Main Group Elements Hemoglobin 8. Chemistry of the Main Group Elements more on that later… 3 8. Chemistry of the Main Group Elements General Trends in Main Group Chemistry Electrical Resistivities: far right: non-metals pnic(t)ogens (pnigo = choke), chalcogens, halogens & noble gases middle: C: Diamond, graphite & fullerenes Si: Silicon, Ge: germanium, Sn & Pb far left: metals alkali metals & alkaline earths: luster, high ability to conduct heat & electricity, malleability 8. Chemistry of the Main Group Elements General Trends in Main Group Chemistry Electrical Resistivities: Carbon conductivity 154.5 pm parallel to layers: σ = C-C 154 pm 2 C=C 134 pm 3 2.6 x 104 sp sp Ω-1cm-1 + pπz T ¼, σ ¿ metal conductivity perp. -

215-216 HH W12-Notes-Ch 15
Chem 215 F12 Notes Notes – Dr. Masato Koreeda - Page 1 of 17. Date: October 5, 2012 Chapter 15: Carboxylic Acids and Their Derivatives and 21.3 B, C/21.5 A “Acyl-Transfer Reactions” I. Introduction Examples: note: R could be "H" R Z R O H R O R' ester O carboxylic acid O O an acyl group bonded to R X R S acid halide* R' an electronegative atom (Z) thioester O X = halogen O R' R, R', R": alkyl, alkenyl, alkynyl, R O R' R N or aryl group R" amide O O O acid anhydride one of or both of R' and R" * acid halides could be "H" R F R Cl R Br R I O O O O acid fluoride acid chloride acid bromide acid iodide R Z sp2 hybridized; trigonal planar making it relatively "uncrowded" O The electronegative O atom polarizes the C=O group, making the C=O carbon "electrophilic." Resonance contribution by Z δ * R Z R Z R Z R Z C C C C O O O δ O hybrid structure The basicity and size of Z determine how much this resonance structure contributes to the hybrid. * The more basic Z is, the more it donates its electron pair, and the more resonance structure * contributes to the hybrid. similar basicity O R' Cl OH OR' NR'R" Trends in basicity: O weakest increasing basiciy strongest base base Check the pKa values of the conjugate acids of these bases. Chem 215 F12 Notes Notes –Dr. Masato Koreeda - Page 2 of 17.