4 M Any-Electron Atoms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Applications of the Variational Monte Carlo Method to the Ground States of the Lithium Atom and Its Ions up to Z = 10 in the Presence of Magnetic Field
Applications of the Variational Monte Carlo Method to the Ground States of the Lithium Atom and its Ions up to Z = 10 in the Presence of Magnetic Field S. B. Doma1), M. O. Shaker2), A. M. Farag2) and F. N. El-Gammal3) 1)Mathematics Department, Faculty of Science, Alexandria University, Egypt E-mail address: [email protected] 2) Mathematics Department, Faculty of Science, Tanta University, Tanta, Egypt 3)Mathematics Department, Faculty of Science, Menofia University, Shebin El-Kom, Egypt Abstract The variational Monte Carlo method is applied to investigate the ground state energy of the lithium atom and its ions up to = 10 in the presence of an external magnetic field regime with = 0 ~ 100 a.u. Our calculations are based on using three forms of compact and accurate trial wave functions, which were put forward in calculating energies in the absence of magnetic field. The obtained results are in good agreement with the most recent accurate values and also with the exact values. Key Words: Atoms in external magnetic field, Variational Monte Carlo method, Lithium atom, Lithium like ions, Total energy. Introduction Over the last decade continuing effort has gone into calculating, with ever increasing accuracy and with various methods, the energies of atoms and ions in neutron star magnetic fields. The motivation comes largely from the fact that features discovered [1–3] in the thermal emission spectra of isolated neutron stars may be due to absorption of photons by heavy atoms in the hot, thin atmospheres of these strongly magnetized cosmic objects [4]. Also, features of heavier elements may be present in the spectra of magnetic white dwarf stars [5, 6]. -
Coordination Chemistry III: Electronic Spectra
160 Chapter 11 Coordination Chemistry III: Electronic Spectra CHAPTER 11: COORDINATION CHEMISTRY III: ELECTRONIC SPECTRA 11.1 a. p3 There are (6!)/(3!3!) = 20 microstates: MS –3/2 –1/2 1/2 3/2 + – – + – + +2 1 1 0 1 1 0 + – – + – + 1 1 –1 1 1 –1 +1 – – + + – + 1 0 0 1 0 0 + – – + + – 1 0 –1 1 0 –1 – – – – + – + – + + + + ML 0 1 0 –1 1 0 –1 1 0 –1 1 0 –1 1– 0– –1+ 1– 0+ –1+ + – – + – + –1 –1 1 –1 –1 1 –1 – – + + – + –1 0 0 –1 0 0 + – – + – + –2 –1 –1 0 –1 –1 0 Terms: L = 0, S = 3/2: 4S (ground state) L = 2, S = 1/2: 2D 2 L = 1, S = 1/2: P 6! 10! b. p1d1 There are 60 microstates: 1!5! 1!9! M S –1 0 1 – – – + + – + + 3 1 2 1 2 , 1 2 1 2 – – + – – + + + 1 1 1 1 , 1 1 1 1 2 + + 0– 2– 0– 2+, 0+ 2– 0 2 1– 0– 1+ 0–, 0+ 1– 1+ 0+ + + 1 0– 1– 1– 0+, 0– 1+ 0 1 + + –1– 2– –1– 2+, –1+ 2– –1 2 – – + – – + + + 1 –1 1 –1 , 1 –1 1 –1 – – – + + – + + ML 0 0 0 0 0 , 0 0 0 0 + + –1– 1– –1+ 1–, –1– 1+ –1 1 – – + – + – + + –1 0 –1 0 , 0 –1 –1 0 – – – + – + + + –1 0 –1 –1 0 , 0 –1 0 –1 – – – + + – 1+ –2+ 1 –2 1 –2 , 1 –2 –1– –1– –1+ –1–, –1– –1+ –1– –1+ –2 + – – + + + 0– –2– 0 –2 , 0 –2 0 –2 –3 –1– –2– –1– –2+, –1+ –2– –1+ –2+ Terms: L = 3, S = 1 3F (ground state) L = 2, S = 0 1D L = 3, S = 0 1F L = 1, S = 1 3P 3 1 L = 2, S = 1 D L = 1, S = 0 P The two electrons have quantum numbers that are independent of each other, because the electrons are in different orbitals. -
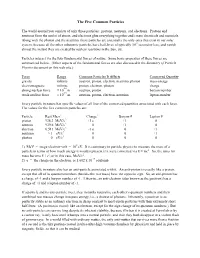
The Five Common Particles
The Five Common Particles The world around you consists of only three particles: protons, neutrons, and electrons. Protons and neutrons form the nuclei of atoms, and electrons glue everything together and create chemicals and materials. Along with the photon and the neutrino, these particles are essentially the only ones that exist in our solar system, because all the other subatomic particles have half-lives of typically 10-9 second or less, and vanish almost the instant they are created by nuclear reactions in the Sun, etc. Particles interact via the four fundamental forces of nature. Some basic properties of these forces are summarized below. (Other aspects of the fundamental forces are also discussed in the Summary of Particle Physics document on this web site.) Force Range Common Particles It Affects Conserved Quantity gravity infinite neutron, proton, electron, neutrino, photon mass-energy electromagnetic infinite proton, electron, photon charge -14 strong nuclear force ≈ 10 m neutron, proton baryon number -15 weak nuclear force ≈ 10 m neutron, proton, electron, neutrino lepton number Every particle in nature has specific values of all four of the conserved quantities associated with each force. The values for the five common particles are: Particle Rest Mass1 Charge2 Baryon # Lepton # proton 938.3 MeV/c2 +1 e +1 0 neutron 939.6 MeV/c2 0 +1 0 electron 0.511 MeV/c2 -1 e 0 +1 neutrino ≈ 1 eV/c2 0 0 +1 photon 0 eV/c2 0 0 0 1) MeV = mega-electron-volt = 106 eV. It is customary in particle physics to measure the mass of a particle in terms of how much energy it would represent if it were converted via E = mc2. -

Review Sheet on Determining Term Symbols
Review Sheet on Determining Term Symbols Term symbols for electronic configurations are useful not only to the spectroscopist but also to the inorganic chemist interested in understanding electronic and magnetic properties of molecules. We will concentrate on the method of Douglas and McDaniel (p. 26ff); however, you may find other treatments equal or superior to the D & M method. Term symbols are a shorthand method used to describe the energy, angular momentum, and spin multiplicity of an atom in any particular state. The general form is a given as Tj where T is a capital letter corresponding to the value of L (the angular momentum quantum number) and may be assigned as S, P, D, F, G, … for |L| = 0, 1, 2, 3, 4, … respectively. The superscript “a” is called the spin multiplicity and can be evaluated as a = 2S +1 where S is the spin quantum number. The subscript “j” is the numerical value of J, a new quantum number defined as: J = l +S, which corresponds to the total orbital and spin angular momentum of the system. The term symbol 3P is read as triplet – Pee state and indicates that there are two unpaired electrons in a state with maximum orbital angular momentum, L=1. The number of microstates (N) of a system corresponds to the total number of distinct arrangements for “e” number of electrons to be placed in “n” number of possible orbital positions. N = # of microstates = n!/(e!(n-e)!) For a set of p orbitals n = 6 since there are 2 positions in each orbital. -

From the Atomic to the Femtoscale Linda Young Argonne National
From the Atomic to the Femtoscale Linda Young Argonne National Laboratory, Argonne, Illinois 60439 Abstract: Atom traps of lithium can be used to provide a new window on few-body atomic and nuclear systems. The trapped atoms form an excellent sample, dense and motionless, for precision measurements. This talk describes experiments using ultracold lithium atoms to study ionization dynamics (a persistent few-body dynamical problem) and outline proposed precision measurements of isotope shifts to determine charge radii of short-lived lithium isotopes (a challenging few-body nuclear physics problem). Introduction The lithium atom, with three electrons and six through eleven nucleons, is a hotbed of activity for few-body theorists in both atomic and nuclear physics. Atomic theorists have the advantage that the forces in the problem, Coulomb interactions, are well known. This advantage simplifies development of many-body techniques for both structure and dynamics [1]. On the atomic physics side, there has been recent effort devoted both to precision calculations of atomic structure [2-5] and to understanding the dynamical correlation between the outgoing electrons in photo triple-ionization [6-9]. On the nuclear physics side, a long-standing program to calculate properties of few-body nuclei has now reached A ≤ 10 [10,11]. In these calculations, the nucleon-nucleon potentials are adjusted to fit the large collection of pp and np scattering data. Impressively, the calculations have fit the binding energies of all A ≤ 10 nuclei to an rms deviation of ≈400 keV, predicted the absence of stable A=5, 8 nuclei [12], and simultaneously predicted rms proton radii, rms neutron radii, quadrupole moments and magnetic moments. -

A Discussion on Characteristics of the Quantum Vacuum
A Discussion on Characteristics of the Quantum Vacuum Harold \Sonny" White∗ NASA/Johnson Space Center, 2101 NASA Pkwy M/C EP411, Houston, TX (Dated: September 17, 2015) This paper will begin by considering the quantum vacuum at the cosmological scale to show that the gravitational coupling constant may be viewed as an emergent phenomenon, or rather a long wavelength consequence of the quantum vacuum. This cosmological viewpoint will be reconsidered on a microscopic scale in the presence of concentrations of \ordinary" matter to determine the impact on the energy state of the quantum vacuum. The derived relationship will be used to predict a radius of the hydrogen atom which will be compared to the Bohr radius for validation. The ramifications of this equation will be explored in the context of the predicted electron mass, the electrostatic force, and the energy density of the electric field around the hydrogen nucleus. It will finally be shown that this perturbed energy state of the quan- tum vacuum can be successfully modeled as a virtual electron-positron plasma, or the Dirac vacuum. PACS numbers: 95.30.Sf, 04.60.Bc, 95.30.Qd, 95.30.Cq, 95.36.+x I. BACKGROUND ON STANDARD MODEL OF COSMOLOGY Prior to developing the central theme of the paper, it will be useful to present the reader with an executive summary of the characteristics and mathematical relationships central to what is now commonly referred to as the standard model of Big Bang cosmology, the Friedmann-Lema^ıtre-Robertson-Walker metric. The Friedmann equations are analytic solutions of the Einstein field equations using the FLRW metric, and Equation(s) (1) show some commonly used forms that include the cosmological constant[1], Λ. -

XSAMS: XML Schema for Atomic, Molecular and Solid Data
XSAMS: XML Schema for Atomic, Molecular and Solid Data Version 0.1.1 Draft Document, January 14, 2011 This version: http://www-amdis.iaea.org/xsams/docu/v0.1.1.pdf Latest version: http://www-amdis.iaea.org/xsams/docu/ Previous versions: http://www-amdis.iaea.org/xsams/docu/v0.1.pdf Editors: M.L. Dubernet, D. Humbert, Yu. Ralchenko Authors: M.L. Dubernet, D. Humbert, Yu. Ralchenko, E. Roueff, D.R. Schultz, H.-K. Chung, B.J. Braams Contributors: N. Moreau, P. Loboda, S. Gagarin, R.E.H. Clark, M. Doronin, T. Marquart Abstract This document presents a proposal for an XML schema aimed at describing Atomic, Molec- ular and Particle Surface Interaction Data in distributed databases around the world. This general XML schema is a collaborative project between International Atomic Energy Agency (Austria), National Institute of Standards and Technology (USA), Universit´ePierre et Marie Curie (France), Oak Ridge National Laboratory (USA) and Paris Observatory (France). Status of this document It is a draft document and it may be updated, replaced, or obsoleted by other documents at any time. It is inappropriate to use this document as reference materials or to cite them as other than “work in progress”. Acknowledgements The authors wish to acknowledge all colleagues and institutes, atomic and molecular database experts and physicists who have collaborated through different discussions to the building up of the concepts described in this document. Change Log Version 0.1: June 2009 Version 0.1.1: November 2010 Contents 1 Introduction 11 1.1 Motivation..................................... 11 1.2 Limitations..................................... 12 2 XSAMS structure, types and attributes 13 2.1 Atomic, Molecular and Particle Surface Interaction Data XML Schema Structure 13 2.2 Attributes .................................... -

Atomic Spectroscopy Summary
ATOMIC SPECTROSCOPY SUMMARY TERM SYMBOLS Because the total angular momentum and energy commute, we can use the angular momentum (L) to label the energy states. Note that electron configurations are ambiguous in that we don’t specify which orbital or which spin each electron has. This means that there are a number of different sets of 푚ℓ and 푚푠 that are consistent with a given configuration. Why are there different energies? Because of electron/electron repulsion and spin-orbit coupling, the ways we can fill the orbitals DO NOT NESCESSARILLY have the same energy. (Remember that we talked about how Hund’s rules come from this. How spin up elelctrons interact less with other spin up electrons in the same subshell.) Components of a Term Symbol: 2푆+1 퐿퐽 RULES FOR TERM SYMBOLS Term Symbols are assigned based on two main rules: 1. Unsöld’s Rule: All filled shells/subshells are ignored when calculating L and S. Basically, all of the electrons will have their contributions to spin and orbital angular momentum canceled by other electrons in the same subshell. 2. “holes”: The state of the “hole” (an empty spot in an orbital where an electron could be but isn’t) is the same as the state of an electron. This means that when we look at a p5 we can pretend it is a p1. This is incredibly useful as it means that instead of 5 electrons to tinker with, you can just act like it is 1. CALCULATING L: L is the Total Angular Momentum L= (sum of ℓ values) ….(smallest positive difference of ℓ values) o 2 s electrons (different shells) L=0+0,…,0-0 L=0 o 1 d electron and 1 p electron L=2+1,…,2-1 L=3, 2, 1 S is the Total Spin S is the total spin quantum number of the electrons in an atom. -

Lesson 1: the Single Electron Atom: Hydrogen
Lesson 1: The Single Electron Atom: Hydrogen Irene K. Metz, Joseph W. Bennett, and Sara E. Mason (Dated: July 24, 2018) Learning Objectives: 1. Utilize quantum numbers and atomic radii information to create input files and run a single-electron calculation. 2. Learn how to read the log and report files to obtain atomic orbital information. 3. Plot the all-electron wavefunction to determine where the electron is likely to be posi- tioned relative to the nucleus. Before doing this exercise, be sure to read through the Ins and Outs of Operation document. So, what do we need to build an atom? Protons, neutrons, and electrons of course! But the mass of a proton is 1800 times greater than that of an electron. Therefore, based on de Broglie’s wave equation, the wavelength of an electron is larger when compared to that of a proton. In other words, the wave-like properties of an electron are important whereas we think of protons and neutrons as particle-like. The separation of the electron from the nucleus is called the Born-Oppenheimer approximation. So now we need the wave-like description of the Hydrogen electron. Hydrogen is the simplest atom on the periodic table and the most abundant element in the universe, and therefore the perfect starting point for atomic orbitals and energies. The compu- tational tool we are going to use is called OPIUM (silly name, right?). Before we get started, we should know what’s needed to create an input file, which OPIUM calls a parameter files. Each parameter file consist of a sequence of ”keyblocks”, containing sets of related parameters. -

1.1. Introduction the Phenomenon of Positron Annihilation Spectroscopy
PRINCIPLES OF POSITRON ANNIHILATION Chapter-1 __________________________________________________________________________________________ 1.1. Introduction The phenomenon of positron annihilation spectroscopy (PAS) has been utilized as nuclear method to probe a variety of material properties as well as to research problems in solid state physics. The field of solid state investigation with positrons started in the early fifties, when it was recognized that information could be obtained about the properties of solids by studying the annihilation of a positron and an electron as given by Dumond et al. [1] and Bendetti and Roichings [2]. In particular, the discovery of the interaction of positrons with defects in crystal solids by Mckenize et al. [3] has given a strong impetus to a further elaboration of the PAS. Currently, PAS is amongst the best nuclear methods, and its most recent developments are documented in the proceedings of the latest positron annihilation conferences [4-8]. PAS is successfully applied for the investigation of electron characteristics and defect structures present in materials, magnetic structures of solids, plastic deformation at low and high temperature, and phase transformations in alloys, semiconductors, polymers, porous material, etc. Its applications extend from advanced problems of solid state physics and materials science to industrial use. It is also widely used in chemistry, biology, and medicine (e.g. locating tumors). As the process of measurement does not mostly influence the properties of the investigated sample, PAS is a non-destructive testing approach that allows the subsequent study of a sample by other methods. As experimental equipment for many applications, PAS is commercially produced and is relatively cheap, thus, increasingly more research laboratories are using PAS for basic research, diagnostics of machine parts working in hard conditions, and for characterization of high-tech materials. -

Bouncing Oil Droplets, De Broglie's Quantum Thermostat And
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 28 August 2018 doi:10.20944/preprints201808.0475.v1 Peer-reviewed version available at Entropy 2018, 20, 780; doi:10.3390/e20100780 Article Bouncing oil droplets, de Broglie’s quantum thermostat and convergence to equilibrium Mohamed Hatifi 1, Ralph Willox 2, Samuel Colin 3 and Thomas Durt 4 1 Aix Marseille Université, CNRS, Centrale Marseille, Institut Fresnel UMR 7249,13013 Marseille, France; hatifi[email protected] 2 Graduate School of Mathematical Sciences, the University of Tokyo, 3-8-1 Komaba, Meguro-ku, 153-8914 Tokyo, Japan; [email protected] 3 Centro Brasileiro de Pesquisas Físicas, Rua Dr. Xavier Sigaud 150,22290-180, Rio de Janeiro – RJ, Brasil; [email protected] 4 Aix Marseille Université, CNRS, Centrale Marseille, Institut Fresnel UMR 7249,13013 Marseille, France; [email protected] Abstract: Recently, the properties of bouncing oil droplets, also known as ‘walkers’, have attracted much attention because they are thought to offer a gateway to a better understanding of quantum behaviour. They indeed constitute a macroscopic realization of wave-particle duality, in the sense that their trajectories are guided by a self-generated surrounding wave. The aim of this paper is to try to describe walker phenomenology in terms of de Broglie-Bohm dynamics and of a stochastic version thereof. In particular, we first study how a stochastic modification of the de Broglie pilot-wave theory, à la Nelson, affects the process of relaxation to quantum equilibrium, and we prove an H-theorem for the relaxation to quantum equilibrium under Nelson-type dynamics. -

5.1 Two-Particle Systems
5.1 Two-Particle Systems We encountered a two-particle system in dealing with the addition of angular momentum. Let's treat such systems in a more formal way. The w.f. for a two-particle system must depend on the spatial coordinates of both particles as @Ψ well as t: Ψ(r1; r2; t), satisfying i~ @t = HΨ, ~2 2 ~2 2 where H = + V (r1; r2; t), −2m1r1 − 2m2r2 and d3r d3r Ψ(r ; r ; t) 2 = 1. 1 2 j 1 2 j R Iff V is independent of time, then we can separate the time and spatial variables, obtaining Ψ(r1; r2; t) = (r1; r2) exp( iEt=~), − where E is the total energy of the system. Let us now make a very fundamental assumption: that each particle occupies a one-particle e.s. [Note that this is often a poor approximation for the true many-body w.f.] The joint e.f. can then be written as the product of two one-particle e.f.'s: (r1; r2) = a(r1) b(r2). Suppose furthermore that the two particles are indistinguishable. Then, the above w.f. is not really adequate since you can't actually tell whether it's particle 1 in state a or particle 2. This indeterminacy is correctly reflected if we replace the above w.f. by (r ; r ) = a(r ) (r ) (r ) a(r ). 1 2 1 b 2 b 1 2 The `plus-or-minus' sign reflects that there are two distinct ways to accomplish this. Thus we are naturally led to consider two kinds of identical particles, which we have come to call `bosons' (+) and `fermions' ( ).