Implications for GI, Immune, and Neuronal Function in Autism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Association Between Maternal Use of Folic Acid Supplements and Risk of Autism Spectrum Disorders in Children
ORIGINAL CONTRIBUTION Association Between Maternal Use of Folic Acid Supplements and Risk of Autism Spectrum Disorders in Children ˚ ´ Pal Suren, MD, MPH Importance Prenatal folic acid supplements reduce the risk of neural tube defects Christine Roth, MSc in children, but it has not been determined whether they protect against other neu- Michaeline Bresnahan, PhD rodevelopmental disorders. Margaretha Haugen, PhD Objective To examine the association between maternal use of prenatal folic acid supplements and subsequent risk of autism spectrum disorders (ASDs) (autistic disor- Mady Hornig, MD der, Asperger syndrome, pervasive developmental disorder–not otherwise specified Deborah Hirtz, MD [PDD-NOS]) in children. Kari Kveim Lie, MD Design, Setting, and Patients The study sample of 85 176 children was derived from the population-based, prospective Norwegian Mother and Child Cohort Study W. Ian Lipkin, MD (MoBa). The children were born in 2002-2008; by the end of follow-up on March 31, Per Magnus, MD, PhD 2012, the age range was 3.3 through 10.2 years (mean, 6.4 years). The exposure of Ted Reichborn-Kjennerud, MD, PhD primary interest was use of folic acid from 4 weeks before to 8 weeks after the start of pregnancy, defined as the first day of the last menstrual period before conception. Synnve Schjølberg, MSc Relative risks of ASDs were estimated by odds ratios (ORs) with 95% CIs in a logistic George Davey Smith, MD, DSc regression analysis. Analyses were adjusted for maternal education level, year of birth, and parity. Anne-Siri Øyen, PhD Main Outcome Measure Specialist-confirmed diagnosis of ASDs. Ezra Susser, MD, DrPH Results At the end of follow-up, 270 children in the study sample had been diag- Camilla Stoltenberg, MD, PhD nosed with ASDs: 114 with autistic disorder, 56 with Asperger syndrome, and 100 with PDD-NOS. -

Lower Autism Risk with Folic Acid Supplements in Pregnancy 12 February 2013
Lower autism risk with folic acid supplements in pregnancy 12 February 2013 Women who took folic acid supplements in early of folic acid supplements in the mother during pregnancy almost halved the risk of having a child pregnancy and a reduced risk of childhood autism. with autism. Beginning to take folic acid supplements later in pregnancy did not reduce the "The study does not prove that folic acid risk. This is shown in new findings from the ABC supplements can prevent childhood autism. Study and Norwegian Mother and Child Cohort However, the findings are so apparent that they Study published in the Journal of The American constitute a good argument to further examine Medical Association (JAMA). possible causal mechanisms. It should also be ascertained whether folic acid is associated with a Women who took folic acid supplements from four reduced risk of other brain disorders in children," weeks before conception to eight weeks into says Surén. pregnancy had a 40 per cent lower risk of giving birth to children with childhood autism (classic Emphasises the importance of folic acid autism). Use of folic acid supplements midway supplements through pregnancy (week 22) had no effect. The results support the Norwegian Directorate of The findings only apply to a lower risk of childhood Health's recommendations for folic acid autism, the most severe form of autism. The supplements during pregnancy and emphasise the results show no reduction in the risk of atypical or importance of starting early—preferably before unspecific autism. The study also investigated the conception. prevalence of Asperger syndrome, but the number of examined children was too low to give a reliable Method result. -

Environmental Health Issue
FIFTH EDITION 2006, Volume 45 R5 Environmental Health and Autism In thIs Issue: Time To GeT a Grip By martha r. Herbert, m.D., ph.D. Beyond Behavior—Biomedical Diagnoses in Autism spectrum Disorders By Margaret L. Bauman, M.D. transforming the Public Debate on neurotoxicants By Elise Miller, M.Ed. ADVERTISEMENT ADVERTISEMENT Autism does not have to be a life sentence You’re not about to give up on your child. Neither Are We. Since , the Autism Treatment Center of America™ has provided innovative training programs for parents and professionals caringifor children challenged by Autism Spectrum Disorders and related developmental difficulties. • Practical Tools • Powerful Results • Limitless Hope c Help your child improve in all areas of over p learning, development, communication and hoto skill acquisition. : © W I Join us for our internationally-acclaimed ll T ERR Son-Rise Program® Start-Up, a y comprehensive weeklong training program for parents and professionals. We don’t put limits on the possibilities for your child. Free 25-Minute Initial Call 877-766-7473 We’ll give you the keys to unlock their world. HOME OF THE SON-RISE PROGRAM® SINCE 1983 South Undermountain Road Sheffield, MA - USA Telephone: -- • E-mail: [email protected] www.autismtreatment.com Copyright © 2006 by The Option Institute & Fellowship. All rights reserved. 02.06-6 CONTENTS December 2006 page 18 SpOTlIGHT Time to Get A Grip By marTHa r. HerBerT, m.D., pH.D. Does an environmental role in autism make sense? How do we decide? And if environment is involved in autism, what do we do about it? These are challenging questions. -

Sensitivity and Specificity of Early Screening for Autism
BJPsych Open (2019) 5, e41, 1–8. doi: 10.1192/bjo.2019.34 Sensitivity and specificity of early screening for autism Pål Surén, Alexandra Saasen-Havdahl, Michaeline Bresnahan, Deborah Hirtz, Mady Hornig, Catherine Lord, Ted Reichborn-Kjennerud, Synnve Schjølberg, Anne-Siri Øyen, Per Magnus, Ezra Susser, W. Ian Lipkin and Camilla Stoltenberg Background (95% CI 58–79) for ASD without phrase speech and 34% (95% CI – Early identification and diagnosis is beneficial for children with 29 40) for ASD with phrase speech. Specificity was then reduced – autism spectrum disorder (ASD). Universal early screening is to 89% (95% CI 89 90). recommended by many experts, but disputed because evidence is limited, and sensitivity and specificity in general populations Conclusions are largely unknown. Early ASD screening with a parent checklist had low sensitivity. It identified mainly individuals with ASD with significant develop- Aims mental delay and captured very few children with ASD with To estimate the sensitivity and specificity of early population- cognitive skills in the normal range. Increasing sensitivity was not based screening for ASDs. possible without severely compromising specificity. Method Declaration of interest The study was based on the Norwegian Mother and Child Cohort C.L. receives royalty for the Social Communication Study. The 36-month cohort questionnaire included the Social Questionnaire, which she has co-authored. Communication Questionnaire (SCQ), a 40-item screening instrument for ASD. Keywords Results Autistic spectrum disorders; developmental disorders; A total of 58 520 mothers (58%) responded to the questionnaire. epidemiology. By the end of follow-up on 31 December 2015, 385 (0.7%) indi- viduals with ASD had been identified among the responders’ Copyright and usage children. -

Prenatal Exposure to Antipsychotic Medication Does Not Increase Odds of Autism, ADHD
Spectrum | Autism Research News https://www.spectrumnews.org NEWS Prenatal exposure to antipsychotic medication does not increase odds of autism, ADHD BY PETER HESS 20 AUGUST 2021 Listen to this story: Children born to mothers who take antipsychotic medications during pregnancy do not have elevated odds of autism or attention deficit hyperactivity disorder (ADHD), nor are they more likely to be born preterm or underweight, according to a study released this past Monday in JAMA Internal Medicine. Some women with schizophrenia, Tourette syndrome or bipolar disorder take antipsychotic drugs, such as aripiprazole, haloperidol or risperidone. Clinicians have long debated whether women should discontinue these medications during pregnancy out of concern for the drugs’ effects on the developing fetus. But children born to mothers who take antipsychotics during pregnancy and to those who do not take them have similar outcomes, the new work shows. “Our findings do not support a recommendation for women to discontinue their regular antipsychotic treatment during pregnancy,” says senior investigator Kenneth Man, research fellow at the University College London School of Pharmacy in the United Kingdom. Prescribing antipsychotics during pregnancy can help prevent potentially dangerous psychotic episodes and ensure that an expectant mother can take care of herself, says Mady Hornig, associate professor of epidemiology at Columbia University, who was not involved in the study. “We certainly don’t want to be cavalier about the use of any medication during pregnancy, but one 1 / 2 Spectrum | Autism Research News https://www.spectrumnews.org also wants to balance out the implications of not treating.” Any apparent effects of antipsychotics on a developing fetus are likely due to the condition being treated, rather than the treatment, the study shows. -
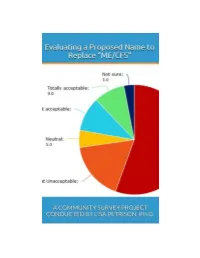
Read the Full Survey Report
Evaluating a Proposed Name to Replace ‘ME/CFS’: A Community Survey Project Conducted by Lisa Petrison, Ph.D. Published by Paradigm Change (March 2015) Copyright 2015 www.paradigmchange.me 2 Table of Contents Executive Summary ...................................................................................................................................... 5 Part 1 – Results Overview .......................................................................................................................... 13 Part 2 - Implications ................................................................................................................................... 29 Part 3 – Survey Questions .......................................................................................................................... 49 Part 4 – Main Survey Results ..................................................................................................................... 66 Part 5 – ME vs. Non-ME Results ............................................................................................................... 110 Part 6 – US ME vs. Non-ME Results ......................................................................................................... 127 Part 7 – Related Illnesses Results ............................................................................................................ 161 Part 8 – US vs. Non-US Results ................................................................................................................. 181 -

Association of MTHFR Gene Variants with Autism
Association of MTHFR Gene Variants with Autism Marvin Boris, M.D.; Allan Goldblatt, P.A.; Clinically available testing for methylenetetrahydrofolate Joseph Galanko, Ph.D.; S. Jill James, Ph.D. reductase (MTHFR) gene mutations (polymorphisms) has recently become available and had been incorporated into our evaluation ABSTRACT process for developmentally delayed children. The MTHFR gene codes for an essential enzyme in folate metabolism. To further Autism is a complex neurodevelopment disorder with understand this condition, we retrospectively evaluated our numerous possible genetic and environmental influences. findings regarding the genomic variations in the gene. MTHFR We retrospectively examined the laboratory data of 168 children enzyme catalyzes the reduction of 5,10-methylenetetrahydrofolate sequentially referred to our facility with a confirmed diagnosis of to 5-methyltetrahydrofolate. Methyltetrahydrofolate is essential in autism or pervasive developmental disabilities (PDD). Since folate one-carbon-donor metabolism for the remethylation of and methylation (single carbon metabolism) are vital in homocysteine to methionine and the generation of metabolically neurological development, we routinely screened children for the active tetrahydrofolate in the methionine synthase reaction.1 common mutations of the methylenetetrahydrofolate reductase Common polymorphisms in the MTHFR gene have been gene (MTHFR), which regulates this pathway. All children had associated with reduced enzyme activity. A detailed review of polymerase chain reaction -

Absence of Evidence for Bornavirus Infection in Schizophrenia, Bipolar
Molecular Psychiatry (2012) 17, 486–493 & 2012 Macmillan Publishers Limited All rights reserved 1359-4184/12 www.nature.com/mp ORIGINAL ARTICLE Absence of evidence for bornavirus infection in schizophrenia, bipolar disorder and major depressive disorder M Hornig1,2, T Briese1,2, J Licinio3, RF Khabbaz4, LL Altshuler5, SG Potkin6, M Schwemmle7, U Siemetzki1, J Mintz5, K Honkavuori1, HC Kraemer8, MF Egan9, PC Whybrow5, WE Bunney6 and WI Lipkin1,2 1Center for Infection and Immunity, Columbia University Mailman School of Public Health, New York, NY, USA; 2Department of Epidemiology, Columbia University Mailman School of Public Health, New York, NY, USA; 3John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia; 4Centers for Disease Control and Prevention, Atlanta, GA, USA; 5Department of Psychiatry and Biobehavioral Sciences, University of California Los Angeles, Los Angeles, CA, USA; 6University of California Irvine, Irvine, CA, USA; 7Department of Virology, Universita¨tsklinikum, Freiburg, Germany; 8Department of Psychiatry and Behavioral Sciences, Stanford University, Palo Alto, CA, USA and 9Clinical Neuroscience, Merck & Company, North Wales, PA, USA In 1983, reports of antibodies in subjects with major depressive disorder (MDD) to an as-yet uncharacterized infectious agent associated with meningoencephalitis in horses and sheep led to molecular cloning of the genome of a novel, negative-stranded neurotropic virus, Borna disease virus (BDV). This advance has enabled the development of new diagnostic assays, including in situ hybridization, PCR and serology based on recombinant proteins. Since these assays were first implemented in 1990, more than 80 studies have reported an association between BDV and a wide range of human illnesses that include MDD, bipolar disorder (BD), schizophrenia (SZ), anxiety disorder, chronic fatigue syndrome, multiple sclerosis, amyo- trophic lateral sclerosis, dementia and glioblastoma multiforme. -

2 James Mif06sun Metabollic Biomarkers
Pathologic Implications of Low Glutathione Levels And Oxidative Stress in Children with Autism: Metabolic Biomarkers and Genetic Polymorphisms S. Jill James, Ph.D. Department of Pediatrics Arkansas Children’s Hospital Research Institute University of Arkansas for Medical Sciences Little Rock, AR Overview Pathways of Folate/Methionine/Glutathione metabolism; Impact of Oxidative Stress Mechanisms and Functions of Glutathione Depletion of glutathione with Thimerosal in vitro Abnormal Methylation and Oxidative Stress in Autistic Children: Treatment with Methyl B-12, Folinic Acid, and TMG Selected Genetic Polymorphisms Associated with the Abnormal Metabolic Profile in Autism Implications of Oxidative Stress in the Pathogenesis of Autism Methionine Transsulfuration to Cysteine and Glutathione Methionine THF 5,10-CH 2THF 1 MS B12 MTHFR 5-CH 3THF Homocysteine B6 THF: tetrahydrofolate Enzymes Methionine Transsulfuration to Cysteine and Glutathione Methionine THF 5,10-CH 2THF 1 MS B12 MTHFR 5-CH 3THF Homocysteine B6 THF: tetrahydrofolate Enzymes Methionine Transsulfuration to Cysteine and Glutathione Methionine Methylation THF SAM Potential (SAM/SAH) MTase Cell Methylation 5,10-CH 2THF 1 MS BHMT 2 B12 SAH MTHFR Betaine 5-CH 3THF SAHH Choline Adenosine Homocysteine B6 THF: tetrahydrofolate Enzymes Methionine Transsulfuration to Cysteine and Glutathione Methionine Methylation THF SAM Potential (SAM/SAH) MTase Cell Methylation 5,10-CH 2THF 1 MS BHMT 2 B12 SAH MTHFR Betaine 5-CH 3THF SAHH Choline Adenosine Homocysteine B6B6 CBS Cystathionine 3 B6 Antioxidant -

Autism Made Easy Therapeutic Nutritional Strategies in the Autistic Spectrum of Disorders
Autism Made Easy Therapeutic Nutritional Strategies in the Autistic Spectrum of Disorders Peta Cohen, MS., RD. Star Academy Johannesburg, Nov 2011 www. PetaCohen .com Therapeutic Nutritional Strategies in ASD Defining the Problem • Autism is treatable, recovery is possible • Epigenetic Phenomenon (methylation) • Complex – Whole Body, Multi-System, Metabolic Disorder – Many targets for simultaneous treatments • Biological Heterogeneity • Chronic Dynamic Encephalopathy • Disorder that Affects the Brain www. PetaCohen .com 2 Autism- A Whole Body, Multi-System, Metabolic Disorder Systems Oxidative Stress Inflammation Methylation Transulfuration Mitochondrial Redoxstatus Function Biotransformation Excitation SNPs – Single Nucleotide Polymorphisms www. PetaCohen .com 3 Autism- A Whole Body, Multi-System, Metabolic Disorder Nervous System Gastro-Intestinal System Respiratory System Immune System Endocrine System Eco- System/Microbiome Cardiovascular System Oxidative Stress Impaired Methylation Inflammation Impaired Trans-Sulfuration Excitation Impaired Detoxification Mitochondrial Dysfunction Nutrient Insufficiencies/Excess pH Balance/Redox Status Cerebral Folate Deficiency SNP – Single Nucleotide Polymorphisms, Epigenetics Genomics Proteomics Metabolomics www. PetaCohen .com 4 Autism- A Whole Body, Multi-System, Metabolic Disorder Stressors Function Feed Forward Dysfunction - Systemic & Metabolic genetics immunity GI biotransformation www. PetaCohen .com 5 Stress Multiple Routes of Exposure – Environmental – Metabolic • Chemical/Toxic • Nutritional -

Doubt Greets Reports of Suramin's Promise for Treating Autism
Spectrum | Autism Research News https://www.spectrumnews.org NEWS Doubt greets reports of suramin’s promise for treating autism BY HANNAH FURFARO 15 JUNE 2017 A drug normally used to treat African sleeping sickness had only mild side effects in a widely reported trial of 10 boys with autism. But many researchers question the trial’s rationale and caution that serious side effects may emerge in larger studies. Half the boys in the trial received a single low dose of the drug, called suramin, intravenously. All five developed a mild rash but experienced no other health problems, researchers reported in May in the Annals of Clinical and Translational Neurology1. But suramin is not a benign medication. In higher doses, it is known to cause anemia and problems with the adrenal gland, which makes hormones, including the stress hormone cortisol. “If I had someone with [autism] in my family, I would certainly not immediately jump to using this as a treatment,” says David Sulzer, professor of psychiatry, neurology and pharmacology at Columbia University. “It easily could be that a sizeable fraction of people who might take this in the future develop additional side effects, which could be bad side effects.” The boys who received the drug showed modest improvements in autism features relative to the five boys in the control group. However, the study was not designed to assess the drug’s effectiveness. The rash may have tipped off the parents as to whether their child was in the treatment or control group — and may have contributed to a placebo effect. -

Center for Solutions for ME/CFS W
Center for Solutions for ME/CFS W. Ian Lipkin, MD Director, Center for Infection and Immunity John Snow Professor of Epidemiology, Mailman School of Public Health Professor of Neurology and Pathology, Vagelos College of Physicians and Surgeons ME/CFS Population 21,736-65,000 ME/CFS patients in NYS (2.6% of US population) 836,000-2,500,000 ME/CFS patients in the US Estimated 84-91% have not yet been diagnosed Estimates of 17-24 million ME/CFS patients worldwide https://www.cdc.gov/me-cfs/about/index.html https://ammes.org/how-many-people-have-mecfs/ The Burden of ME/CFS DIAGNOSING AND TREATING MYALGIC ENCEPHALOMYELITIS/ CHRONIC FATIGUE SYNDROME (ME/CFS) - U.S. ME/CFS CLINICIAN COALITION - Version 2 ∙ July 2020 ME/CFS studies have shown one peak of onset between ages 11-19 and a second between 30-39. At least 25% of patients are bedbound or housebound and up to 75% are unable to work or attend school. Symptoms can persist for years, and most patients never regain their pre-disease functioning ME/CFS costs the US $17-$24 billion annually in lost productivity and direct medical costs. https://drive.google.com/file/d/1SG7hlJTCSDrDHqvioPMq-cX-rgRKXjfk/view What is Known About ME/CFS Prior Infections KEY FACTS ∙ FEBRUARY 2015 Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Epstein-Barr virus What are the symptoms and other effects of ME/CFS? Ross River virus 11% develop ME/CFS symptoms • Reduction or impairment in ability to carry out normal daily activities, Coxiella burnetti accompanied by profound fatigue • Post-exertional malaise