Information to Users
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
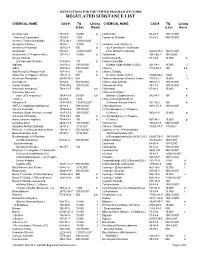
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Diborane (B2H6) 2 Heavier Boron Hydrides in the Form of Volatile Liquids Such As
Even in the absence of contamination, diborane is so reactive that it will slowly decompose by itself, forming H gas and Diborane (B2H6) 2 heavier boron hydrides in the form of volatile liquids such as Storage and Delivery B5H9 and sublimable solids such as B10H14. These heavier hydrides are delivered with the gas and can cause Air Products supplies a wide range of gases and chemicals contamination of the process and fouling of the delivery system that are used for integrated circuit, flat panel display and components. Figure 1 shows the internals of the first stage of photovoltaic device manufacturing. Understanding the proper a two-stage gas regulator that was fouled by B10H14 deposits. storage and handling requirements of these specialty materials is essential to obtain consistent results and to ensure the highest level of process safety. In this technical bulletin, we will provide the user with some of the knowledge required for its effective storage and use. Figure 1: Picture of the first stage of diborane mixture regulator Diborane (formula B2H6) is a colorless, spontaneously fouled with solid decomposition products. flammable (pyrophoric) gas. It burns rapidly in air and reacts vigorously with water with the evolution of heat. The gas has The gas-phase decomposition rate of diborane was shown to a distinct, noxious odor and is extremely toxic by inhalation or increase with concentration and with temperature. Pure by skin exposure. Because of these hazards, it is essential diborane is so reactive in its pure state that it may only be that all gas handling systems used for diborane be tightly stored or transported if surrounded with dry ice to prevent rapid sealed and carefully leak checked. -
![A Calix[4]Arene Based Boronic Acid Catalyst for Amide Bond Formation: Proof of Principle Study](https://docslib.b-cdn.net/cover/2183/a-calix-4-arene-based-boronic-acid-catalyst-for-amide-bond-formation-proof-of-principle-study-122183.webp)
A Calix[4]Arene Based Boronic Acid Catalyst for Amide Bond Formation: Proof of Principle Study
The Free Internet Journal Paper for Organic Chemistry Archive for Arkivoc 2018, part v, 221-229 Organic Chemistry A calix[4]arene based boronic acid catalyst for amide bond formation: proof of principle study Asslly Tafara Mafaune and Gareth E. Arnott* Department of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1, Matieland, South Africa Email: [email protected] Received 01-23-2018 Accepted 04-14-2018 Published on line 06-25-2018 Abstract A calix[4]arene boronic acid was synthesized and tested for catalysis in amide formation. The results were positive and paved the way for future designs, even though protodeboronation was observed under the conditions employed. Keywords: Amide bond catalysis, calix[4]arene boronic acid, protodeboronation DOI: https://doi.org/10.24820/ark.5550190.p010.492 Page 221 ©ARKAT USA, Inc Arkivoc 2018, v, 221-229 Mafaune, A. T. et al. Introduction The importance of amide bonds to human kind cannot be overemphasized; they are ubiquitous in nature and indispensable in chemical applications. They can be found in compounds such as the polymers that make our lives easier; the insecticides and agrochemicals that ensure that we have food on our tables; and most notably in the pharmaceutical drugs that help us live longer. Amide bonds are also part of the building blocks of biological systems, linking together amino acid units forming peptides, proteins and enzymes. The amide is arguably the most important functional group in chemistry and it also happens to be the most frequently synthesized in medicinal chemistry.1 Because of the versatility and importance of the amide bond, catalytic direct amide formation has been highlighted as a top priority reaction from a green chemistry viewpoint. -

Phenyl Replacement Reactions: Solvent Effects on Reactions of Boroxines with Primary Amines
Phenyl Replacement Reactions: Solvent Effects on Reactions of Boroxines with Primary Amines A thesis presented to the faculty of the Graduate School of Western Carolina University in partial fulfillment of the requirements for the degree of Master of Science in Chemistry. By Nicholas John Wilcox Director: Dr. William R. Kwochka Associate Professor of Chemistry Chemistry Department Committee Members: Dr. Channa De Silva, Chemistry Dr. Brian Dinkelmeyer, Chemistry March 2015 TABLE OF CONTENTS Page List of Figures ......................................................................................................................... iii List of Schemes ...................................................................................................................... vi Abstract................................................................................................................................... vii Chapter One: Introduction ....................................................................................................... 1 1.1 Background ...................................................................................................... 1 1.2 Complexes created by dative bonds ................................................................ 3 Chapter Two: Results and Discussion ..................................................................................... 7 2.1 Displacement and Complexation Reactions ..................................................... 7 Chapter Three: Conclusion .................................................................................................... -

Safety Data Sheet
SAFETY DATA SHEET Revision Date 27-February-2020 Revision Number 2 1. Identification Product Name Potassium hydride, 30% w/w in mineral oil Cat No. : L13266 Synonyms No information available Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Importer/Distributor Manufacturer Fisher Scientific Alfa Aesar 112 Colonnade Road, Thermo Fisher Scientific Chemicals, Inc. Ottawa, ON K2E 7L6, 30 Bond Street, Ward Hill, MA 01835-8099 Canada Tel: 800-343-0660 Fax: 800-322-4757 Tel: 1-800-234-7437 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (800) 579-7421. 2. Hazard(s) identification Classification WHMIS 2015 Classification Classified as hazardous under the Hazardous Products Regulations (SOR/2015-17) Substances/mixtures which, in contact with water, emit Category 1 flammable gases Skin Corrosion/Irritation Category 1 B Serious Eye Damage/Eye Irritation Category 1 Specific target organ toxicity (single exposure) Category 3 Target Organs - Respiratory system. Aspiration Toxicity Category 1 Physical Hazards Not Otherwise Classified Category 1 Reacts violently with water Label Elements Signal Word Danger Hazard Statements In contact with water releases flammable gases which may ignite spontaneously ______________________________________________________________________________________________ Page 1 / 7 Potassium hydride, 30% w/w in mineral oil Revision Date 27-February-2020 ______________________________________________________________________________________________ May be fatal if swallowed and enters airways Causes severe skin burns and eye damage May cause respiratory irritation Reacts violently with water Precautionary Statements Manufacturer Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. -

Page 1 of 26 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 26 RSC Advances The ternary amide KLi 3(NH 2)4: an important intermediate in the potassium compounds-added Li –N–H systems Bao-Xia Dong, Liang Song, Jun Ge, Yun-Lei Teng*, Shi-Yang Zhang College of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, P. R. China. In this paper, the KH-added LiH–NH 3, KH-added LiH–LiNH 2, KH-added LiNH 2, and KNH 2-added LiNH 2 systems were systematically investigated. It was found that the ternary amide KLi 3(NH 2)4 was an important intermediate that was inclined to be formed in the dehydrogenation and hydrogenation processes of the potassium compounds-added Li–N–H system. -

1 Structure, Properties, and Preparation of Boronic Acid Derivatives Overview of Their Reactions and Applications Dennis G
j1 1 Structure, Properties, and Preparation of Boronic Acid Derivatives Overview of Their Reactions and Applications Dennis G. Hall 1.1 Introduction and Historical Background Structurally, boronic acids are trivalent boron-containing organic compounds that possess one carbon-based substituent (i.e., a CÀB bond) and two hydroxyl groups to fill the remaining valences on the boron atom (Figure 1.1). With only six valence electrons and a consequent deficiency of two electrons, the sp2-hybridized boron atom possesses a vacant p-orbital. This low-energy orbital is orthogonal to the three substituents, which are oriented in a trigonal planar geometry. Unlike carbox- ylic acids, their carbon analogues, boronic acids, are not found in nature. These abiotic compounds are derived synthetically from primary sources of boron such as boric acid, which is made by the acidification of borax with carbon dioxide. Borate esters, one of the key precursors of boronic acid derivatives, are made by simple dehydration of boric acid with alcohols. The first preparation and isolation of a boronic acid was reported by Frankland in 1860 [1]. By treating diethylzinc with triethylborate, the highly air-sensitive triethylborane was obtained, and its slow oxidation in ambient air eventually provided ethylboronic acid. Boronic acids are the products of a twofold oxidation of boranes. Their stability to atmospheric oxidation is considerably superior to that of borinic acids, which result from the first oxidation of boranes. The product of a third oxidation of boranes, boric acid, is a very stable and relatively benign compound to humans (Section 1.2.2.3). Their unique properties and reactivity as mild organic Lewis acids, coupled with their stability and ease of handling, are what make boronic acids a particularly attractive class of synthetic intermediates. -

276262828.Pdf
FUNDAMENTAL STUDIES OF CATALYTIC DEHYDROGENATION ON ALUMINA-SUPPORTED SIZE-SELECTED PLATINUM CLUSTER MODEL CATALYSTS by Eric Thomas Baxter A dissertation submitted to the faculty of The University of Utah in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Chemistry The University of Utah May 2018 Copyright © Eric Thomas Baxter 2018 All Rights Reserved T h e U n iversity of Utah Graduate School STATEMENT OF DISSERTATION APPROVAL The dissertation of Eric Thomas Baxter has been approved by the following supervisory committee members: Scott L. Anderson , Chair 11/13/2017 Date Approved Peter B. Armentrout , Member 11/13/2017 Date Approved Marc D. Porter , Member 11/13/2017 Date Approved Ilya Zharov , Member 11/13/2017 Date Approved Sivaraman Guruswamy , Member 11/13/2017 Date Approved and by Cynthia J. Burrows , Chair/Dean of the Department/College/School of Chemistry and by David B. Kieda, Dean of The Graduate School. ABSTRACT The research presented in this dissertation focuses on the use of platinum-based catalysts to enhance endothermic fuel cooling. Chapter 1 gives a brief introduction to the motivation for this work. Chapter 2 presents fundamental studies on the catalytic dehydrogenation of ethylene by size-selected Ptn (n = 4, 7, 8) clusters deposited onto thin film alumina supports. The model catalysts were probed by a combination of experimental and theoretical techniques including; temperature-programmed desorption and reaction (TPD/R), low energy ion scattering spectroscopy (ISS), X-ray photoelectron spectroscopy (XPS), plane wave density-functional theory (PW-DFT), and statistical mechanical theory. It is shown that the Pt clusters dehydrogenated approximately half of the initially adsorbed ethylene, leading to deactivation of the catalyst via (coking) carbon deposition. -

Minutesbasel Approved
Approved minutes, Basel 2018 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Approved minutes for Division Committee Meeting Basel, Switzerland, 13–14 August, 2018 1. Welcome, introductory remarks and housekeeping announcements Alan Hutton (ATH) welcomed everybody to the meeting, extending a special welcome to those who were attending the Division Committee (DC) meeting for the first time, and noting the presence of three former Presidents of the Division, as well as the current Secretary General of IUPAC. He described the house rules and arrangements for the course of the meeting. 2. Attendance and apologies Present: Alan T. Hutton (President, ATH), Karl-Heinz Hellwich (Past-President, KHH), Risto S. Laitinen (Secretary, RSL), Michael A. Beckett (MAB), Edwin C. Constable (ECC), Ture Damhus (TD), Richard M. Hartshorn (RMH), Elisabeth Mansfield (EM), Gerry P. Moss (GPM), Michelle M. Rogers (MMR), Molly A. Strausbaugh (MAS), Clare Tovee (CT), Andrey Yerin (AY) (see also Appendix 1) Apologies: Fabio Aricò (FA), Maria Atanasova Petrova (MA), Neil Burford (NB), (JC), Ana Maria da Costa Ferreira (ACF), Safiye Erdem (SE), Rafał Kruszyński (RK), Robin Macaluso (RM), , Ladda Meesuk (LM), Ebbe Nordlander (EN), Amelia P. Rauter (APR), Erik Szabó (ES), Keith T. Taylor (KTT), Jiří Vohlídal (JV) Invited observer: G. Jeffery Leigh (GJL) No replies: József Nagy, Sangho Koo 3. Introduction of attendees A short round of introductions was made. A new titular member, Prof. Edwin C. Constable and a new Associate Member, Dr. Clare Tovee, were attending the meeting of the Division Committee for the first time in this function. KHH informed the meeting that the following persons had passed away during recent years: Peter A. -

Identification of Diborane(4) with Bridging B–H–B Bonds
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Identification of diborane(4) with bridging B–H–B bonds† Cite this: Chem. Sci.,2015,6, 6872 Sheng-Lung Chou,a Jen-Iu Lo,a Yu-Chain Peng,a Meng-Yeh Lin,a Hsiao-Chi Lu,a Bing-Ming Cheng*a and J. F. Ogilvieb The irradiation of diborane(6) dispersed in solid neon at 3 K with tunable far-ultraviolet light from a Received 17th July 2015 synchrotron yielded a set of IR absorption lines, the pattern of which implies a carrier containing two Accepted 14th August 2015 boron atoms. According to isotope effects and quantum-chemical calculations, we identified this new DOI: 10.1039/c5sc02586a species as diborane(4), B2H4, possessing two bridging B–H–B bonds. Our work thus establishes a new www.rsc.org/chemicalscience prototype, diborane(4), for bridging B–H–B bonds in molecular structures. Introduction The B–H–B linkage in these compounds is considered to be an atypical electron-decient covalent chemical bond.7 Creative Commons Attribution 3.0 Unported Licence. The structure that B2H6 adopts in its electronic ground state According to calculations, diborane species possessing less and most stable conformation contains two bridging B–H–B than 6 hydrogen atoms and bridging B–H–Bbondscan 8–13 bonds, and four terminal B–H bonds. Although Bauer, from exist, but all possible candidates are transient species, experiments with the electron diffraction of gaseous samples in difficult to prepare and to identify. Since the pioneering work the laboratory of Pauling and Brockway, favoured a structure of involving absorption spectra of samples at temperatures less 14,15 diborane(6) with a central B–B bond analogous to that of than 100 K, many free radicals and other unstable 16–20 ethane,1 Longuet-Higgins deduced the bridged structure for compounds dispersed in inert hosts have been detected. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine