Identification and Characterization of Plant-Derived Alkaloids, Corydine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Network Pharmacology and Traditional Chinese Medicine: Devel- Opment of Anti-Diabetic Therapies Zhongxia Lu1, Wenjun Xu1, Xi Chen2, Changyu Li3* and Yitao Chen1*
ISSN: 2377-3634 Lu et al. Int J Diabetes Clin Res 2017, 4:077 DOI: 10.23937/2377-3634/1410077 Volume 4 | Issue 2 International Journal of Open Access Diabetes and Clinical Research REVIEW ARTICLE Network Pharmacology and Traditional Chinese Medicine: Devel- opment of Anti-Diabetic Therapies Zhongxia Lu1, Wenjun Xu1, Xi Chen2, Changyu Li3* and Yitao Chen1* 1College of Life Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China 2College of Traditional Chinese Medicine, Beijing University of Chinese Medicine, China Check for 3College of Pharmacy, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China updates *Corresponding author: Yitao Chen, MD, College of Life Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China, E-mail: [email protected]; Changyu Li, MD, College of Pharmacy, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, China, E-mail: [email protected] Abstract Research and Development Dilemma for Anti- Diabetes Drugs Partly due to the failure of single-target drugs, diabetes mel- litus, a chronic metabolic disease with complex pathogene- Type 2 Diabetes Mellitus (T2DM), generally agreed sis and long-term medication requirements, is increasing in to be caused by insulin resistance and/or insulin defi- prevalence worldwide and urgently needs multi-component and multi-target treatments. Traditional Chinese herbs are ciency, constitutes almost 95 percent of all diabetes the principal drug of Chinese medicine, which is effective cases [4]. Because of the pathogenesis, the mainstre- against diabetes. However, Chinese herbs’ mechanism of am anti-diabetic drugs are insulin secretagogues (sul- action is difficult to elucidate due to its multiple components phonylureas and meglitinide analogues), insulin sensi- and multi-target effects. -

Supplementary Materials 1
Supplementary materials 1 Table S1 The characteristics of botanical preparations potentially containing alkenylbenzenes on the Chinese market. Botanical Pin Yin Name Form Ingredients Recommendation for daily intake (g) preparations (汉语) Plant food supplements (PFS) Si Ji Kang Mei Yang Xin Yuan -Rou Dou Kou xylooligosaccharide, isomalt, nutmeg (myristica PFS 1 Fu He Tang Pian tablet 4 tablets (1.4 g) fragrans), galangal, cinnamon, chicken gizzards (四季康美养心源-肉豆蔻复合糖片) Ai Si Meng Hui Xiang fennel seed, figs, prunes, dates, apples, St.Johns 2-4 tablets (2.8-5.6 g) PFS 2 Fu He Pian tablet Breed, jamaican ginger root (爱司盟茴香复合片) Zi Ran Mei Xiao Hui Xiaong Jiao Nang foeniculi powder, cinnamomi cortex, papaya PFS 3 capsule concentrated powder, green oat concentrated powder, 3 capsules (1.8 g) (自然美小茴香胶囊) brewer’s yeast, cabbage, monkey head mushroom An Mei Qi Hui Xiang Cao Ben Fu He Pian fennel seed, perilla seed, cassia seed, herbaceous PFS 4 tablet 1-2 tablets (1.4-2.8 g) (安美奇茴香草本复合片) complex papaya enzymes, bromelain enzymes, lactobacillus An Mei Qi Jiao Su Xian Wei Ying Yang Pian acidophilus, apple fiber, lemon plup fiber, fennel PFS 5 tablet seed, cascara sagrada, jamaican ginger root, herbal 2 tablets (2.7 g) (安美奇酵素纤维营养片) support complex (figs, prunes, dates, apples, St. Johns bread) Table S1 (continued) The characteristics of botanical preparations potentially containing alkenylbenzenes on the Chinese market. Pin Yin Name Botanical Form Ingredients Recommendation for daily intake (g) preparations (汉语) Gan Cao Pian glycyrrhiza uralensis, licorice -

Anti-Cholinergic Alkaloids As Potential Therapeutic Agents for Alzheimer's Disease
Indian Journal of Biochemistry & Biophysics Vol. 50, April 2013, pp. 120-125 Anti-cholinergic alkaloids as potential therapeutic agents for Alzheimer’s disease: An in silico approach Huma Naaz, Swati Singh, Veda P Pandey, Priyanka Singh and Upendra N Dwivedi* Bioinformatics Infrastructure Facility, Center of Excellence in Bioinformatics, Department of Biochemistry, University of Lucknow, Lucknow 226 007, India Received 10 September 2012; revised 25 January 2013 Alzheimer’s disease (AD), a progressive neurodegenerative disorder with many cognitive and neuropsychiatric symptoms is biochemically characterized by a significant decrease in the brain neurotransmitter acetylcholine (ACh). Plant-derived metabolites, including alkaloids have been reported to possess neuroprotective properties and are considered to be safe, thus have potential for developing effective therapeutic molecules for neurological disorders, such as AD. Therefore, in the present study, thirteen plant-derived alkaloids, namely pleiocarpine, kopsinine, pleiocarpamine (from Pleiocarpa mutica, family: Annonaceae), oliveroline, noroliveroline, liridonine, isooncodine, polyfothine, darienine (from Polyalthia longifolia, family: Apocynaceae) and eburnamine, eburnamonine, eburnamenine and geissoschizol (from Hunteria zeylanica, family: Apocynaceae) were analyzed for their anti-cholinergic action through docking with acetylcholinesterase (AChE) as target. Among the alkaloids, pleiocarpine showed promising anti-cholinergic potential, while its amino derivative showed about six-fold -

Effect of Wine and Vinegar Processing of Rhizoma Corydalis on the Tissue Distribution of Tetrahydropalmatine, Protopine and Dehydrocorydaline in Rats
Michigan Technological University Digital Commons @ Michigan Tech Michigan Tech Publications 1-18-2012 Effect of wine and vinegar processing of Rhizoma Corydalis on the tissue distribution of tetrahydropalmatine, protopine and dehydrocorydaline in rats Zhiying Dou Tianjin University of Traditional Chinese Medicine Kefeng Li Michigan Technological University Ping Wang Tianjin University of Traditional Chinese Medicine Liu Cao Tianjin University of Traditional Chinese Medicine Follow this and additional works at: https://digitalcommons.mtu.edu/michigantech-p Part of the Biology Commons Recommended Citation Dou, Z., Li, K., Wang, P., & Cao, L. (2012). Effect of wine and vinegar processing of Rhizoma Corydalis on the tissue distribution of tetrahydropalmatine, protopine and dehydrocorydaline in rats. Molecules, 17(1), 951-970. http://doi.org/10.3390/molecules17010951 Retrieved from: https://digitalcommons.mtu.edu/michigantech-p/1969 Follow this and additional works at: https://digitalcommons.mtu.edu/michigantech-p Part of the Biology Commons Molecules 2012, 17, 951-970; doi:10.3390/molecules17010951 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Effect of Wine and Vinegar Processing of Rhizoma Corydalis on the Tissue Distribution of Tetrahydropalmatine, Protopine and Dehydrocorydaline in Rats Zhiying Dou 1,*, Kefeng Li 2, Ping Wang 1 and Liu Cao 1 1 College of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin 300193, China 2 Department of Biological Sciences, Michigan Technological University, Houghton, MI 49931, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel./Fax: +86-22-5959-6235. Received: 29 November 2011; in revised form: 5 January 2012 / Accepted: 9 January 2012 / Published: 18 January 2012 Abstract: Vinegar and wine processing of medicinal plants are two traditional pharmaceutical techniques which have been used for thousands of years in China. -

NRDC: Generally Recognized As Secret
NRDC Report April 2014 Generally Recognized as Secret: Chemicals Added to Food in the United States Tom Neltner, J.D., Maricel Maffini, Ph.D. Natural Resources Defense Council In April 2014, the Natural Resources Defense Council (NRDC) released a report raising concerns about a loophole in the Food Additives Amendment of 1958 for substances designated by food manufacturers as “generally recognized as safe” (GRAS). The report identified 56 companies that appeared to market 275 chemicals for use in food based on undisclosed GRAS safety determinations. For each chemical we identified in this study, we did not find evidence that FDA had cleared them for use in food. The 1958 law exempted from the formal, extended FDA approval process common food ingredients like vinegar and vegetable oil whose use qualifies as GRAS. It may have appeared reasonable at the time, but that exemption has been stretched into a loophole that has swallowed the law. The exemption allows manufacturers to make safety determinations that the uses of their newest chemicals in food are safe without notifying the FDA. The agency’s attempts to limit these undisclosed GRAS determinations by asking industry to voluntarily inform the FDA about their chemicals are insufficient to ensure the safety of our food in today’s global marketplace with a complex food supply. Furthermore, no other developed country in the world has a system like GRAS to provide oversight of food ingredients. In Table 1 and 2 of the report, NRDC identified the 56 companies and the number of chemicals that each company appeared to market as GRAS without FDA clearance. -
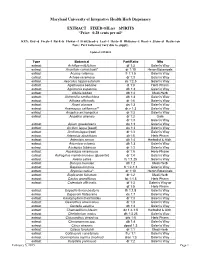
SPIRITS *Price: 0.28 Cents Per Ml*
Maryland University of Integrative Health Herb Dispensary EXTRACT FIXED OIL(s) SPIRITS *Price: 0.28 cents per ml* KEY: Dry=d Fresh=f Bark=b Flower=f Fruit/Seed=s Leaf=l Herb=H Rhizome=z Root=r Stem=st Resin=rsn Note: Part ratio may vary due to supply. Updated 1/29/2015 Type Botanical Part/Ratio Mfg extract Achillea millefolium df 1:3 Galen’s Way extract Aconitum carmichaeli* dr 1:10 Heron Botanicals extract Acorus calamus fr 1:1.5 Galen’s Way extract Actaea racemosa dr 1:3 Galen’s Way extract Aesculus hippocastanum ds 1:2.5 Galen’s Way extract Agathosma betulina dl 1:3 Herb Pharm extract Agrimonia eupatoria dh 1:3 Galen’s Way extract Albizia lebbek db 1:2 Medi-Herb extract Alchemilla xanthochlora dh 1:3 Galen’s Way extract Althaea officinalis dr 1:6 Galen’s Way extract Ammi visnaga ds 1:3 Galen’s Way extract Anemopsis californica** dr,z 1:3 Galen’s Way extract Angelica archangelica dr 1:3 Galen’s Way extract Angelica sinensis dr 1:2 Gaia dr 1:3 Galen’s Way extract Apium graveoloens ds 1:3 Galen’s Way extract Arctium lappa (seed) ds 1:3 Galen’s Way extract Arctium lappa (root) dr 1:3 Galen’s Way extract Artemisia absinthium dh1:5 Herb Pharm extract Artemisia annua dh 1:4 Herbalist & Alch. extract Artemisia vulgaris dh 1:3 Galen’s Way extract Asclepias tuberosa dr 1:3 Galen’s Way extract Asparagus racemosus dr 1:5 Herb-Pharm extract Astragalus membranaceus (glycerite) dr 1:4 Galen’s Way extract Avena sativa fs 1:1.25 Galen’s Way extract Bacopa monnieri dh 1:2 Medi-Herb extract Baptisia tinctoria fr 1:2-1:3 Galen’s Way extract Bryonia cretica* dr 1:10 Heron Botanicals extract Bupleurum falcatum dr 1:2 Medi-Herb extract Cactus grandiflorus fst 1:1.5 Herb Pharm extract Calendula officinalis df 1:3 Galen’s Way or df 1:5 Herb Pharm extract Capsella bursa-pastoris fh 1:1.5 Galen’s Way extract Capsicum frutescens ds 1:7 Galen’s Way extract Caulophyllum thalictroides dr 1:3 Galen’s Way extract Ceanothus americanus dr 1:3 Galen’s Way extract Centella asiatica dh 1:2 Galen’s Way extract Chamaelirium luteum dz 1:4-1:5 Herbalist & Alch. -

5994392 Tion of Application No. 67375.734 Eb3-1685, PEN. T
USOO5994392A United States Patent (19) 11 Patent Number: 5,994,392 Shashoua (45) Date of Patent: Nov.30, 1999 54 ANTIPSYCHOTIC PRODRUGS COMPRISING 5,120,760 6/1992 Horrobin ................................. 514/458 AN ANTIPSYCHOTICAGENT COUPLED TO 5,141,958 8/1992 Crozier-Willi et al. ................ 514/558 AN UNSATURATED FATTY ACID 5,216,023 6/1993 Literati et al. .......................... 514/538 5,246,726 9/1993 Horrobin et al. ....................... 424/646 5,516,800 5/1996 Horrobin et al. ....................... 514/560 75 Inventor: Victor E. Shashoua, Brookline, Mass. 5,580,556 12/1996 Horrobin ................................ 424/85.4 73 Assignee: Neuromedica, Inc., Conshohocken, Pa. FOREIGN PATENT DOCUMENTS 30009 6/1981 European Pat. Off.. 21 Appl. No.: 08/462,820 009 1694 10/1983 European Pat. Off.. 22 Filed: Jun. 5, 1995 09 1694 10/1983 European Pat. Off.. 91694 10/1983 European Pat. Off.. Related U.S. Application Data 59-025327 2/1984 Japan. 1153629 6/1989 Japan. 63 Continuation of application No. 08/080,675, Jun. 21, 1993, 1203331 8/1989 Japan. abandoned, which is a continuation of application No. 07/952,191, Sep. 28, 1992, abandoned, which is a continu- (List continued on next page.) ation of application No. 07/577,329, Sep. 4, 1990, aban doned, which is a continuation-in-part of application No. OTHER PUBLICATIONS 07/535,812,tion of application Jun. 11, No. 1990, 67,375.734 abandoned, Eb3-1685, which is a continu-PEN. T. Higuchi et al. 66 Prodrugs as Noye Drug Delivery Sys 4,933,324, which is a continuation-in-part of application No. -

Charles University in Prague, Faculty of Pharmacy in Hradec Kralove Department of Pharmaceutical Botany and Ecology DIPLOMA TH
Charles University in Prague, Faculty of Pharmacy In Hradec Kralove Department of Pharmaceutical Botany and Ecology ______________________________________________________________________ DIPLOMA THESIS Biological Activity of Plant Metabolites XVII. Alkaloids of Corydalis yanhusuo W.T. Wang Supervisor of Diploma Work: Assoc. Prof. RNDr. Lubomir Opletal, CSc. Head of Department: Prof. RNDr. Luděk Jahodář, CSc. Hradec Králové April, 2011 Gabriella Cipra Declaration I declare that this thesis is my original copyrighted work. All literature and other sources from which I extracted my research in the process are listed in the bibliography and all work is properly cited. This work has not been used to gain another or same title. Acknowledgements I wish to express my deepest gratitude, first and foremost to Assoc. Prof. RNDr. Lubomír Opletal, CSc. for all his guidance, support and enthusiasm with the preparation of this thesis. I would also like to thank the Department of Pharmaceutical Botany and Ecology for the pleasant working environment, as well as Assoc. Prof. PharmDr. Jiří Kuneš, Ph.D. for preparation and interpretation of NMR spectra, Ing. Kateřina Macáková for biological activity measurements, and Ing. Lucie Cahlíková, Ph.D. for MS spectra measurements and interpretations. This work was financially supported by Specific University Research Foundation No SVV-2011-263002 (Study of biologically active compounds in prespective of their prevention and treatment in civil diseases) Table of Contents 1 INTRODUCTION 7 2 AIM OF WORK 10 3 THEORETICAL -

Analysis of L-Dopa Induced Dyskinesias in 51 Patients with Parkinsonism
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.34.6.668 on 1 December 1971. Downloaded from J. Neurol. Neurosurg. Psychiat., 1971, 34, 668-673 Analysis of L-dopa induced dyskinesias in 51 patients with Parkinsonism R. J. MONES, T. S. ELIZAN, AND G. J. SIEGEL From the Department of Neurology, Mount Sinai Medical School, New York, New York, U.S.A. SUMMARY An analysis of 51 patients with Parkinsonism who have developed L-dopa induced dyskinesias is presented. The cause has not been proven, although various hypotheses are discussed. One third of the total number of patients treated developed dyskinesia. These patients tend to re- spond better to L-dopa than the other group. There is a tendency for the older patient or the patient with long-standing disease to develop dyskinesias. There appears to be no way of predicting which patients will develop dyskinesia by analysis of the symptoms or the aetiology of the Parkinsonism syndrome. The unilateral characteristic of the dyskinesia in patients with hemi-Parkinsonism and patients with unilateral thalamotomies suggests that structural abnormalities are critical in deter-guest. Protected by copyright. mining the presence and localization of dyskinesias. This is supported by non-occurrence of similarly treated patients without Parkinsonism. L-dopa is now established as the treatment of choice and 45 started in the hospital. The protocol for the medical for Parkinsonism. Many articles have appeared in and neurological evaluation of these patients, the follow- the literature since the initial work of Cotzias, up care, and the laboratory data are included in a pre- Van vious paper (Mones, 1969). -

Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Sedative
Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Sedative Chemical Dosage (+)-BORNYL-ISOVALERATE -- (-)-DICENTRINE LD50=187 1,8-CINEOLE -- 2-METHYLBUT-3-ENE-2-OL -- 6-GINGEROL -- 6-SHOGAOL -- ACYLSPINOSIN -- ADENOSINE -- AKUAMMIDINE -- ALPHA-PINENE -- ALPHA-TERPINEOL -- AMYL-BUTYRATE -- AMYLASE -- ANEMONIN -- ANGELIC-ACID -- ANGELICIN ED=20-80 ANISATIN 0.03 mg/kg ANNOMONTINE -- APIGENIN 30-100 mg/kg ARECOLINE 1 mg/kg ASARONE -- ASCARIDOLE -- ATHEROSPERMINE -- BAICALIN -- BALDRINAL -- BENZALDEHYDE -- BENZYL-ALCOHOL -- Chemical Dosage BERBERASTINE -- BERBERINE -- BERGENIN -- BETA-AMYRIN-PALMITATE -- BETA-EUDESMOL -- BETA-PHENYLETHANOL -- BETA-RESERCYCLIC-ACID -- BORNEOL -- BORNYL-ACETATE -- BOSWELLIC-ACID 20-55 mg/kg ipr rat BRAHMINOSIDE -- BRAHMOSIDE -- BULBOCAPNINE -- BUTYL-PHTHALIDE -- CAFFEIC-ACID 500 mg CANNABIDIOLIC-ACID -- CANNABINOL ED=200 CARPACIN -- CARVONE -- CARYOPHYLLENE -- CHELIDONINE -- CHIKUSETSUSAPONIN -- CINNAMALDEHYDE -- CITRAL ED 1-32 mg/kg CITRAL 1 mg/kg CITRONELLAL ED=1 mg/kg CITRONELLOL -- 2 Chemical Dosage CODEINE -- COLUBRIN -- COLUBRINOSIDE -- CORYDINE -- CORYNANTHEINE -- COUMARIN -- CRYOGENINE -- CRYPTOCARYALACTONE 250 mg/kg CUMINALDEHYDE -- CUSSONOSIDE-A -- CYCLOSTACHINE-A -- DAIGREMONTIANIN -- DELTA-9-THC 10 mg/orl/man/day DESERPIDINE -- DESMETHOXYANGONIN 200 mg/kg ipr DIAZEPAM 40-200 ug/lg/3-4x/day DICENTRINE LD50=187 DIDROVALTRATUM -- DIHYDROKAWAIN -- DIHYDROMETHYSTICIN 60 mg/kg ipr DIHYDROVALTRATE -- DILLAPIOL ED50=1.57 DIMETHOXYALLYLBENZENE -- DIMETHYLVINYLCARBINOL -- DIPENTENE -

Proprietary Products Guide
Proprietary Formulas Our proprietary blends are unlike any on the market and offer acute relief as well as tonic support for constitutional health. Highly effective and accessibly priced, our remedies inspire repeat purchases and loyal following. Our practitioner partners love having these on hand, and find they are well matched for many common health concerns. All our formulas are available for distribution under your label and are also offered bulk. Commitment to Quality Superior products are only possible with superior medicinals. We source from the most reputable herb suppliers, including local farms and responsible wildcrafters. We are proud to work only with Chinese herb distributors who employ rigid standards and quality assurance testing, and continually offer us better access to organically grown Chinese medicinals. All of the Western herbs we buy are certified organic, locally grown, or ethically wildcrafted. Each of our vendors goes through our GMP supplier qualification program, and each herb is analyzed against our rigorous specifications to ensure the identity, purity, and potency of every medicinal. We are registered to produce certified organic products by CCOF. Our Approach Launched by a professional herbalist / acupuncturist, and a medical doctor, Five Flavors Herbs bridges the therapeutic traditions of East and West. We combine scientific research and clinical practice with millenniums old theory of healing, offering reliable tools for practitioners and purveyors. Truly unique formulations, our extract line stands out both in approach and effectiveness. Nervous System Remedies Elation (Xiao Yao San +) ~ mood support Lifts the mood and promotes a healthy emotional response to stress and monthly hormonal changes.* Take 15-60 drops in ¼ cup water 3-5 times daily or as directed. -

Diversity of the Mountain Flora of Central Asia with Emphasis on Alkaloid-Producing Plants
diversity Review Diversity of the Mountain Flora of Central Asia with Emphasis on Alkaloid-Producing Plants Karimjan Tayjanov 1, Nilufar Z. Mamadalieva 1,* and Michael Wink 2 1 Institute of the Chemistry of Plant Substances, Academy of Sciences, Mirzo Ulugbek str. 77, 100170 Tashkent, Uzbekistan; [email protected] 2 Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Im Neuenheimer Feld 364, 69120 Heidelberg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +9-987-126-25913 Academic Editor: Ipek Kurtboke Received: 22 November 2016; Accepted: 13 February 2017; Published: 17 February 2017 Abstract: The mountains of Central Asia with 70 large and small mountain ranges represent species-rich plant biodiversity hotspots. Major mountains include Saur, Tarbagatai, Dzungarian Alatau, Tien Shan, Pamir-Alai and Kopet Dag. Because a range of altitudinal belts exists, the region is characterized by high biological diversity at ecosystem, species and population levels. In addition, the contact between Asian and Mediterranean flora in Central Asia has created unique plant communities. More than 8100 plant species have been recorded for the territory of Central Asia; about 5000–6000 of them grow in the mountains. The aim of this review is to summarize all the available data from 1930 to date on alkaloid-containing plants of the Central Asian mountains. In Saur 301 of a total of 661 species, in Tarbagatai 487 out of 1195, in Dzungarian Alatau 699 out of 1080, in Tien Shan 1177 out of 3251, in Pamir-Alai 1165 out of 3422 and in Kopet Dag 438 out of 1942 species produce alkaloids. The review also tabulates the individual alkaloids which were detected in the plants from the Central Asian mountains.