Amplicon Sequencing of the 16S-ITS-23S Rrna Operon with Long-Read Technology for Improved Phylogenetic Classification of Uncultured Prokaryotes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Structure of the Perennial Growth of Disa Un/Flora Berg
THE STRUCTURE OF THE PERENNIAL GROWTH OF DISA UN/FLORA BERG. ( ORCHIDACEAE) HONOURS SYSTEMATICS PROJECT JANET THOMAS OCTOBER 1990 SUPERVISOR: DR . .H.P. LINDER University of Cape Town The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgement of the source. The thesis is to be used for private study or non- commercial research purposes only. Published by the University of Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University of Cape Town BOLUS LIBRARY 1 ABSTRACT The perennation of orchids is poorly understood, in particular that of the Orchidoidae. The understanding of perennation in the Orchidoidae is important because the root-stem tuberoid .is used as the one character defining the Orchidoidae as a monophyletic group. The root-stem tuberoid has never been examined for variation before. This project focuses on perennial growth in the Diseae in order to study the structbre and function of the root stem tuberoid in relation tp other organs and to contribute to the understanding of Orchidoid phylogeny. , INTRODUCTION Host te1perate monocotyledons have evolved underground resting or perennating organs for the climatically unfavourable season (Holttum 1955). A period of underground existence may allow a plant to escape unfavourable conditions, to counter environmental uncertainty, and to build reserves for flowering episodes (Calvo 1990). This is especially evident in the temperate members of the Orchidaceae and is made possible through sympodial growth· (Withnerj1974). Not .all temperate orchids have a resting period although they do have sympodial growth and do perennate. -

PLANT LIST 2019 DISA DISA Uniflora £15 Orange
DAVE PARKINSON PLANTS PLANT LIST 2019 DISA DISA uniflora £15 Orange. Large flowered. Beautiful species. DISA uniflora-pink £15 Later flowering species. Large flowers of pale dusky pink. DISA uniflora-Carmine £15 Large flowers of attractive carmine. Later flowering species, very scarce DISA uniflora-Red River £15 Large red flowers. Short growing species. DISA aurata £15 Many small golden yellow flowers on a long stem. Small plant, species. DISA tripetaloides £15 Many small white/pink flowers on a long stem arising from a small plant species. In short supply.(Sold Out) HYBRIDS DISA Brides Dream £15 Veitchii x Foam. DISA Child Safety Transvaal £15 Unifoam x Betty's Bay DISA Child Safety Transvaal Sonia' £15 Large flowered, tall growing. Brilliant cerise pink DISA Collette Cywes ‘Blush’ £15 Tall growing. Very pale pink. In short supply DISA Constantia £12 Kewdiot x Betty's Bay DISA Diores £12 Pretty orange/red unnamed clone DISA Diores-Inca Warrior £12 Red/orange flowers on a robust plant. Excellent choice to start a collection DISA Diores-Inca Princess £12 Tall growing. Large pale pink flowers DISA Diores-Inca City £12 Pink slightly later flowering DISA Diores Inca Gold £12 Deep pink, despite the name DISA Diores `Inca King` £12 Large flowered Pink DISA Diorosa £12 Very tall growing. Large flowered pink DISA Foam 'Zoe' £15 Tall growing, large orangy flowers with pink overlay, petal tips pink DISA Foam £12 Mainly orange, Large Flowers DISA Glasgow Orchid Conference £15 Large flowers of orange or red. Strong plant. Short growing DISA Kalahari Sands £15 Foam x Unifoam DISA Kalahari Sands 'Tina' £15 Selected clone. -

Disa Orchids and Their Ex Vitro Seed Culture©
124 Combined Proceedings International Plant Propagators’ Society, Volume 58, 2008 Disa Orchids and Their Ex Vitro Seed Culture© Ken Davey 211 Ngamotu Rd, New Plymouth Email: [email protected] INTRODUCTION Before I retired I was employed as Technical Officer in The Fernery in Pukekura Park, New Plymouth. Part of my job was to look after the very diverse orchid col- lection that the Fernery is known for. This extensive collection has been assembled over many years by George Fuller and others. This paper will look at some of the background in the development of the terrestrial orchid species Disa uniflora and its hybrids as export cut flower and pot plant crops in New Zealand (N.Z.). THE PLANTS Disa is a genus of over 100 species of terrestrial orchids that mainly occur in south- ern Africa in habitats that range from swamp to seasonally dry areas. Many of the species have small flowers and are of collector value only. They can also be a real challenge to grow ex situ. Disa uniflora, also known as “Pride of Table Mountain,” is centred on the Table Mountain area, where it grows in damp and or other seasonally wet sites such as stream sides or swamps and can even be found peeping out from under waterfalls. Although the species name is “uniflora” because the first plant described only had a single flower on each stem, other plants were later found to be multiflowered. They can have quite large flowers that range from red/orange to orange/gold with the oc- casional gold/yellow. -

A New Species of Disa (Orchidaceae) from Mpumalanga, South Africa ⁎ D
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector South African Journal of Botany 72 (2006) 551–554 www.elsevier.com/locate/sajb A new species of Disa (Orchidaceae) from Mpumalanga, South Africa ⁎ D. McMurtry a, , T.J. Edwards b, B. Bytebier c a Whyte Thorne, P O Box 218, Carino 1204, South Africa b School of Biological and Conservation Sciences, University of KwaZulu–Natal Pietermaritzburg, Private Bag X01, Scottsville 3209, South Africa c Biochemistry Department, Stellenbosch University, Private Bag X1, Stellenbosch 7602, South Africa Received 10 November 2005; accepted 8 March 2006 Abstract A new species, Disa vigilans D. McMurtry and T.J. Edwards, is described from the Mpumalanga Escarpment. The species is a member of the Disa Section Stenocarpa Lindl. Its alliances are discussed in terms of its morphology and its phylogenetic placement is elucidated using molecular data. D. vigilans has previously been considered as an anomalous form of Disa montana Sond. but is more closely allied to Disa amoena H.P. Linder. © 2006 SAAB. Published by Elsevier B.V. All rights reserved. Keywords: Disa; Draensberg endemic; New species; Orchidaceae; Section Stenocarpae; South Africa; Mpumalanga province 1. Introduction with 3 main veins, margins thickened and translucent. Inflorescence lax, cylindrical, 40–75 mm long; bracts light green suffused pinkish Disa is the largest genus of Orchidaceae in southern Africa (162 with darker green veins, linear-lanceolate, acuminate, 11–29×2– spp.) and has been the focus of considerable taxonomic investigation 3 mm, scarious at anthesis. Flowers white suffused with carmine- (Linder, 1981a,b, 1986; Linder and Kurzweil, 1994). -

Sotwp 2016.Pdf
STATE OF THE WORLD’S PLANTS OF THE WORLD’S STATE 2016 The staff and trustees of the Royal Botanic Gardens, Kew and the Kew Foundation would like to thank the Sfumato Foundation for generously funding the State of the World’s Plants project. State of the World’s Plants 2016 Citation This report should be cited as: RBG Kew (2016). The State of the World’s Plants Report – 2016. Royal Botanic Gardens, Kew ISBN: 978-1-84246-628-5 © The Board of Trustees of the Royal Botanic Gardens, Kew (2016) (unless otherwise stated) Printed on 100% recycled paper The State of the World’s Plants 1 Contents Introduction to the State of the World’s Plants Describing the world’s plants 4 Naming and counting the world’s plants 10 New plant species discovered in 2015 14 Plant evolutionary relationships and plant genomes 18 Useful plants 24 Important plant areas 28 Country focus: status of knowledge of Brazilian plants Global threats to plants 34 Climate change 40 Global land-cover change 46 Invasive species 52 Plant diseases – state of research 58 Extinction risk and threats to plants Policies and international trade 64 CITES and the prevention of illegal trade 70 The Nagoya Protocol on Access to Genetic Resources and Benefit Sharing 76 References 80 Contributors and acknowledgments 2 Introduction to the State of the World’s Plants Introduction to the State of the World’s Plants This is the first document to collate current knowledge on as well as policies and international agreements that are the state of the world’s plants. -
AEDC Hypersonic Tests to Benefit from Improvements to Emissivity Measurement Accuracy
PRSRT STD US POSTAGE PAID TULLAHOMA TN Vol. 67, No. 24 Arnold AFB, Tenn. PERMIT NO. 29 December 21, 2020 AEDC Base Civil Engineering Branch vital to mission success at Arnold AFB By Jill Pickett ety of capabilities and servic- AEDC Public Affairs es that directly support every person, every organization From mowing grass to op- and every test capability on erating test utility systems, Arnold AFB,” said John Lavi- the Arnold Engineering De- olette, TSDC chief. “Many velopment Complex Civil En- of these services are founda- gineering Branch, or TSDC, tional to successfully accom- ensures the readiness of the plishing the installation’s test grounds, facilities and utility mission.” systems at Arnold Air Force A team of DOD and con- Base, headquarters of AEDC. tractor personnel work to- “The Test Support Di- gether to sustain, restore and Garry Sudberry, an Arnold Engineering Development Complex fuel farm machinist, sets up vision’s Civil Engineering the A Plant Fuel System, Sept. 10, at Arnold Air Force Base. The AEDC Base Civil Engineering Branch oversees utilities at Arnold AFB, including jet fuel for testing needs. (U.S. Air Force Branch provides a wide vari- See SUCCESS, page 4 photo by Jill Pickett) AEDC hypersonic tests to benefit from improvements to emissivity measurement accuracy By Jill Pickett but there are limitations on the AEDC Public Affairs number and locations these can be placed. Infrared cameras al- As hypersonic vehicle de- low for continuous measure- velopment heats up, the Ar- ment of the surface tempera- nold Engineering Development tures of a model under test, but Complex Aerothermal Measure- the infrared camera measure- ments Laboratory (ATML) is ments must be verified first. -

Orchidoideae: Orchidaceae) Author(S): H
The Phylogeny and Classification of the Diseae (Orchidoideae: Orchidaceae) Author(s): H. P. Linder and H. Kurzweil Source: Annals of the Missouri Botanical Garden, Vol. 81, No. 4 (1994), pp. 687-713 Published by: Missouri Botanical Garden Press Stable URL: http://www.jstor.org/stable/2399916 Accessed: 27-07-2016 11:10 UTC Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://about.jstor.org/terms JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Missouri Botanical Garden Press is collaborating with JSTOR to digitize, preserve and extend access to Annals of the Missouri Botanical Garden This content downloaded from 137.158.114.36 on Wed, 27 Jul 2016 11:10:19 UTC All use subject to http://about.jstor.org/terms THE PHYLOGENY AND H. P. Linder2 and H. Kurzweil2'3 CLASSIFICATION OF THE DISEAE (ORCHIDOIDEAE: ORCHIDACEAE)l ABSTRACT The subtribal classification of the Diseae (Orchidoideae) is reviewed in light of the available morphological, leaf anatomical, and palynological data. These data are critically assessed, and the more prominent features are illustrated. The data are analyzed cladistically, and the robustness of the various components of the most parsimonious tree is assessed by a bootstrap analysis. Based on the cladistic analysis and the bootstrap analysis, a new classification is proposed for the Diseae. -

Disa Staerkeriana (Orchidaceae): a New Species from Mpumalanga, South Africa
Phytotaxa 203 (2): 192–198 ISSN 1179-3155 (print edition) www.mapress.com/phytotaxa/ PHYTOTAXA Copyright © 2015 Magnolia Press Article ISSN 1179-3163 (online edition) http://dx.doi.org/10.11646/phytotaxa.203.2.9 Disa staerkeriana (Orchidaceae): a new species from Mpumalanga, South Africa DOUGLAS McMURTRY¹ & BENNY BYTEBIER² ¹Whyte Thorne, P.O. Box 218, Karino 1204, South Africa ²School of Life Sciences, University of KwaZulu-Natal, Pr. Bag X01, 3209 Scottsville, South Africa; E-mail: [email protected] Abstract A new orchid species, Disa staerkeriana is described from the Hartebeesvlakte in the Mpumalanga Province of South Africa. It is a member of Disa section Stenocarpa and is affiliated to D. amoena and D. montana. An updated artificial key to Disa section Stenocarpa is provided. Introduction The orchid genus Disa Bergius (1767: 348) (Disinae, Orchideae, Orchidoideae) currently consists of 184 species (Govaerts 2014). It is largely endemic to continental Africa but extends to Madagascar (five species), Réunion (one species) and the Arabian Peninsula (one species). In South Africa, 143 species occur of which 128 are endemic to the country and 88 are endemic to the Cape Floristic Region, its centre of diversity (Galley et al. 2007). Following a molecular phylogenetic analysis (Bytebier et al. 2007a), the segregate genus Schizodium Lindley (1838: 358) was included in Disa and the genus was subdivided into 18 sections (Bytebier et al. 2008). After the publication of the authoritative “Orchids of Southern Africa” (Linder & Kurzweil 1999), six new Disa species have been described from South Africa. Three of these, Disa albomagentea E.G.H.Oliv. -

Use of ITS2 Region As the Universal DNA Barcode for Plants and Animals
Use of ITS2 Region as the Universal DNA Barcode for Plants and Animals Hui Yao1., Jingyuan Song1., Chang Liu1., Kun Luo1,2, Jianping Han1, Ying Li1, Xiaohui Pang1, Hongxi Xu4, Yingjie Zhu3*, Peigen Xiao1, Shilin Chen1* 1 Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, People’s Republic of China, 2 College of Pharmacy, Hubei University of Chinese Medicine, Wuhan, Hubei, People’s Republic of China, 3 School of Bioscience and Engineering, Southwest Jiaotong University, Chengdu, Sichuan, People’s Republic of China, 4 Chinese Medicine Laboratory, Hong Kong Jockey Club Institute of Chinese Medicine, Hong Kong, People’s Republic of China Abstract Background: The internal transcribed spacer 2 (ITS2) region of nuclear ribosomal DNA is regarded as one of the candidate DNA barcodes because it possesses a number of valuable characteristics, such as the availability of conserved regions for designing universal primers, the ease of its amplification, and sufficient variability to distinguish even closely related species. However, a general analysis of its ability to discriminate species in a comprehensive sample set is lacking. Methodology/Principal Findings: In the current study, 50,790 plant and 12,221 animal ITS2 sequences downloaded from GenBank were evaluated according to sequence length, GC content, intra- and inter-specific divergence, and efficiency of identification. The results show that the inter-specific divergence of congeneric species in plants and animals was greater than its corresponding intra-specific variations. The success rates for using the ITS2 region to identify dicotyledons, monocotyledons, gymnosperms, ferns, mosses, and animals were 76.1%, 74.2%, 67.1%, 88.1%, 77.4%, and 91.7% at the species level, respectively. -

Nectar Spur Evolution in the Mexican Lobelias (Campanulaceae:Lobelioideae)1
American Journal of Botany 92(3): 558±562. 2005. NECTAR SPUR EVOLUTION IN THE MEXICAN LOBELIAS (CAMPANULACEAE:LOBELIOIDEAE)1 MARGARET M. KOOPMAN2 AND TINA J. AYERS3 Department of Biological Sciences, P.O. Box 5640, Northern Arizona University, Flagstaff, Arizona 86011-5640 USA Phylogenetic studies are often hampered by the independent evolution of characters that may potentially obscure relationships. The adaptive signi®cance of the nectar spur and its evolution within the Mexican lobeliads (Campanulaceae) is considered here. The taxonomic delimitations of Heterotoma from the Mexican species within the genera Lobelia and Calcaratolobelia were tested. Inde- pendent molecular data were gathered to determine whether the Mexican spurred lobeliads should be treated as distinct genera. The internal transcribed spacer (ITS) region from 18±26S nuclear rDNA and chloroplast DNA from the 39 trnK intron were sequenced from 14 representative species. Our data suggest that Heterotoma, as originally conceived, is a good evolutionary unit within Lobelia and that the presence of a nectar spur is an important morphological character that can be used in de®ning phylogenetic position. This study also suggests that morphological changes associated with hummingbird pollination have evolved more than once in the Mexican lobeliads, from small blue-¯owered, insect-pollinated relatives. Key words: Heterotoma; ITS; Lobelia; nectar spur; pollination; systematics; 39 trnK intron. A fundamental objective in evolutionary biology is the ex- order for successful mutualisms to occur, pollinators need to amination and discovery of the causes and effects of species be both pro®cient in nectar harvesting and effective in polli- origin. The use of phylogenetic data to study the evolution of nation. -

A Molecular Phylogeny Reveals Widespread Floral
Molecular Phylogenetics and Evolution 51 (2009) 100–110 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Pollinators underestimated: A molecular phylogeny reveals widespread floral convergence in oil-secreting orchids (sub-tribe Coryciinae) of the Cape of South Africa Richard J. Waterman a,b,*, Anton Pauw c, Timothy G. Barraclough a, Vincent Savolainen a,b a Division of Biology, Imperial College London, Silwood Park Campus, Ascot, Berkshire SL5 7PY, UK b Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey TW9 3DS, UK c Department of Botany and Zoology, Stellenbosch University, Private Bag X1, Matieland 7602, South Africa article info abstract Article history: The oil-secreting orchids of southern Africa belong to the sub-tribe Coryciinae within Diseae. A phylogeny Received 18 December 2007 of Diseae is inferred using sequence data from all genera in the tribe, with an emphasis on resolving gen- Revised 2 May 2008 eric classifications within Coryciinae. Nuclear (ITS) and plastid (trnLF and matK) gene region sequences Accepted 13 May 2008 were analysed for 79 ingroup taxa and three outgroup taxa. Coryciinae is confirmed to be diphyletic, with Available online 24 May 2008 Disperis and Coryciinae sensu stricto (s.s.) forming separate monophyletic clades. The current genera Cory- cium and Pterygodium are not monophyletic according to our analysis and we propose a subdivision of Keywords: Coryciinae s.s. into 10 monophyletic clades including three monotypic groups. Previous generic classifi- Ceratandra cations of Coryciinae s.s. have been hampered by convergent evolution of floral parts, a consequence of Convergent evolution Corycium few pollinator species and limited pollinia attachment sites in the oil-bee pollination system common Disperis to this group. -
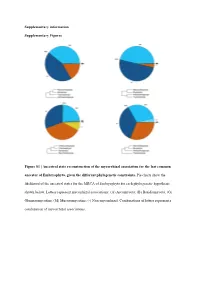
Ancestral State Reconstruction of the Mycorrhizal Association for the Last Common Ancestor of Embryophyta, Given the Different Phylogenetic Constraints
Supplementary information Supplementary Figures Figure S1 | Ancestral state reconstruction of the mycorrhizal association for the last common ancestor of Embryophyta, given the different phylogenetic constraints. Pie charts show the likelihood of the ancestral states for the MRCA of Embryophyta for each phylogenetic hypothesis shown below. Letters represent mycorrhizal associations: (A) Ascomycota; (B) Basidiomycota; (G) Glomeromycotina; (M) Mucoromycotina; (-) Non-mycorrhizal. Combinations of letters represent a combination of mycorrhizal associations. Austrocedrus chilensis Chamaecyparis obtusa Sequoiadendron giganteum Prumnopitys taxifolia Prumnopitys Prumnopitys montana Prumnopitys Prumnopitys ferruginea Prumnopitys Araucaria angustifolia Araucaria Dacrycarpus dacrydioides Dacrycarpus Taxus baccata Podocarpus oleifolius Podocarpus Afrocarpus falcatus Afrocarpus Ephedra fragilis Nymphaea alba Nymphaea Gnetum gnemon Abies alba Abies balsamea Austrobaileya scandens Austrobaileya Abies nordmanniana Thalictrum minus Thalictrum Abies homolepis Caltha palustris Caltha Abies magnifica ia repens Ranunculus Abies religiosa Ranunculus montanus Ranunculus Clematis vitalba Clematis Keteleeria davidiana Anemone patens Anemone Tsuga canadensis Vitis vinifera Vitis Tsuga mertensiana Saxifraga oppositifolia Saxifraga Larix decidua Hypericum maculatum Hypericum Larix gmelinii Phyllanthus calycinus Phyllanthus Larix kaempferi Hieronyma oblonga Hieronyma Pseudotsuga menziesii Salix reinii Salix Picea abies Salix polaris Salix Picea crassifolia Salix herbacea