A Dissertation Entitled Group V Imido Complexes Bearing Mono-Anionic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with Phosphines, Dioxygen and Water
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2005 Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with Phosphines, Dioxygen and Water Laurel Anne Morton University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Chemistry Commons Recommended Citation Morton, Laurel Anne, "Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter- Conversion and Reactions with Phosphines, Dioxygen and Water. " PhD diss., University of Tennessee, 2005. https://trace.tennessee.edu/utk_graddiss/2258 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Laurel Anne Morton entitled "Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with Phosphines, Dioxygen and Water." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. Ziling (Ben) Xue, Major Professor We have read this dissertation and recommend its acceptance: Craig E. Barnes, Charles D. Feigerle, X. Peter Zhang, John R. Collier Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) To the Graduate Council: I am submitting herewith a dissertation written by Laurel Anne Morton entitled "Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. -

Polyphenolic Compounds Extracted and Purified from Buddleja Globosa
molecules Article Polyphenolic Compounds Extracted and Purified from Buddleja Globosa Hope (Buddlejaceae) Leaves Using Natural Deep Eutectic Solvents and Centrifugal Partition Chromatography Jeniffer Torres-Vega 1 , Sergio Gómez-Alonso 2 , José Pérez-Navarro 2 , Julio Alarcón-Enos 3 and Edgar Pastene-Navarrete 1,3,* 1 Laboratorio de Farmacognosia, Departamento de Farmacia, Facultad de Farmacia, Universidad de Concepción, Concepción PC4030000, Chile; [email protected] 2 Regional Institute for Applied Scientific Research, Faculty of Chemical Sciences, University of Castilla-La Mancha, PC13071 Castilla-La Mancha, Spain; [email protected] (S.G.-A.); [email protected] (J.P.-N.) 3 Laboratorio de Síntesis y Biotransformación de Productos Naturales, Universidad del Bío-Bío, Chillán PC3800708, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-(42)-246-3000 Abstract: Chemical profiling of Buddleja globosa was performed by high-performance liquid chro- matography coupled to electrospray ionization (HPLC-DAD-ESI-IT/MS) and quadrupole time-of-flight high-resolution mass spectrometry (HPLC-ESI-QTOF/MS). The identification of 17 main phenolic com- pounds in B. globosa leaf extracts was achieved. Along with caffeoyl glucoside isomers, caffeoylshikimic Citation: Torres-Vega, J.; acid and several verbascoside derivatives (β-hydroxyverbascoside and β-hydroxyisoverbascoside) were Gómez-Alonso, S.; Pérez-Navarro, J.; Alarcón-Enos, J.; Pastene-Navarrete, identified. Among flavonoid compounds, the presence of 6-hydroxyluteolin-7-O-glucoside, quercetin-3- E. Polyphenolic Compounds O-glucoside, luteolin 7-O-glucoside, apigenin 7-O-glucoside was confirmed. Campneoside I, forsytho- Extracted and Purified from Buddleja side B, lipedoside A and forsythoside A were identified along with verbascoside, isoverbascoside, Globosa Hope (Buddlejaceae) Leaves eukovoside and martynoside. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -
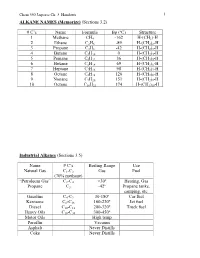
C's Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)
Chem 350 Jasperse Ch. 3 Handouts 1 ALKANE NAMES (Memorize) (Sections 3.2) # C’s Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)-H 2 Ethane C2H6 -89 H-(CH2)2-H 3 Propane C3H8 -42 H-(CH2)3-H 4 Butane C4H10 0 H-(CH2)4-H 5 Pentane C5H12 36 H-(CH2)5-H 6 Hexane C6H14 69 H-(CH2)6-H 7 Heptane C7H16 98 H-(CH2)7-H 8 Octane C8H18 126 H-(CH2)8-H 9 Nonane C9H20 151 H-(CH2)9-H 10 Octane C10H22 174 H-(CH2)10-H Industrial Alkanes (Sections 3.5) Name # C’s Boiling Range Use Natural Gas C1-C3 Gas Fuel (70% methane) “Petroleum Gas” C2-C4 <30º Heating, Gas Propane C3 -42º Propane tanks, camping, etc. Gasoline C4-C9 30-180º Car fuel Kerosene C8-C16 160-230º Jet fuel Diesel C10-C18 200-320º Truck fuel Heavy Oils C16-C30 300-450º Motor Oils High temp Paraffin Vacuum Asphalt Never Distills Coke Never Distills Chem 350 Jasperse Ch. 3 Handouts 2 Nomenclature of Alkanes (Sections 3.3) Systematic IUPAC Rules for Branched and Substituted Alkanes (Section 3.3B) 1. Longest continuous C-chain “core name” 2. Number core chain from an end nearest a substituent 3. Name substituents as “alkyl” groups: 4. Specify the location of substituents using numbers (hyphenate the #’s) • If >2 substituents, list alphabetically • Use di-, tri-, tetra- if the same substituent is repeated. (But ignore these in alphabetizing). Punctuation Notes: • Hyphenate numbers • Do not put a space between substituents and the core name Special Names for Some 3 or 4-carbon Substituents H3C CH3 CH Memorize H3C C H3C CH3 Isopropyl t-butyl or tert-butyl H2 H2 CH3 H3C C H3C C CH H3C CH Others C C C C CH3 H3C C H2 H H H2 2 2 H2 n-propyl n-butyl isobutyl s-butyl (n for "normal") Another Classification System Primary (1º): with one attached carbon Secondary (2º): with two attached carbons Tertiary (3º): with three attached carbons H C C C C C C C C H 1º H 2º C 3º Very Complex Substituents (Not responsible) Substituent: (1-ethyl-2,3-dimethylpentyl) Overall: 9-(1-ethyl-2,3-dimethylpentyl)nonadecane Chem 350 Jasperse Ch. -

Influence of Conjugation Axis on the Optical and Electronic Properties Of
Article pubs.acs.org/joc Influence of Conjugation Axis on the Optical and Electronic Properties of Aryl-Substituted Benzobisoxazoles † ‡ ‡ † † Brian C. Tlach, Aimeé L. Tomlinson, Alden G. Ryno, Dawn D. Knoble, Dana L. Drochner, † Kyle J. Krager, and Malika Jeffries-EL*, † Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States ‡ Department of Chemistry, University of North Georgia, Dahlonega, Georgia 30597, United States *S Supporting Information ABSTRACT: Six different 2,6-diethyl-4,8-diarylbenzo[1,2-d:4,5-d′]bis(oxazoles) and four different 2,4,6,8-tetraarylbenzobisoxazoles were synthesized in two steps: a Lewis acid catalyzed orthoester cyclization followed by a Suzuki or Stille cross-coupling with various arenes. The influence of aryl group substitution and/or conjugation axis variation on the optical and electronic properties of these benzobis(oxazole) (BBO) compounds was evaluated. Structural modifications could be used to alter the HOMO, LUMO, and band gap over a range of 1.0, 0.5, and 0.5 eV, respectively. However, depending on the location and identity of the substituent, the HOMO level can be altered without significantly impacting the LUMO level. This is supported by the calculated frontier molecular orbitals. Our results indicate that the FMOs and band gaps of benzobisoxazoles can be readily modified either jointly or individually. ■ INTRODUCTION Among the aforementioned examples, the benzo[1,2-d:4,5- d′]bis(oxazole) (BBO)-based cruciforms are particularly During the past four decades, interest in the development of π- interesting, since these molecules have two different con- conjugated materials has increased due to their potential use as jugation pathways: 2,6-conjugation through the oxazole rings replacements for inorganic materials in a variety of semi- and 4,8-conjugation through the central benzene ring (Scheme conducting applications including field effect transistors − − 1). -

Alkyne Metathesis Catalysts: Scope and Future André Mortreux, Olivier Coutelier
Alkyne Metathesis Catalysts: Scope And Future André Mortreux, Olivier Coutelier To cite this version: André Mortreux, Olivier Coutelier. Alkyne Metathesis Catalysts: Scope And Future. Journal of Molecular Catalysis A: Chemical, Elsevier, 2006, 254, pp.96-104. 10.1016/j.molcata.2006.03.054. hal-00107451 HAL Id: hal-00107451 https://hal.archives-ouvertes.fr/hal-00107451 Submitted on 18 Oct 2006 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ALKYNE METATHESIS CATALYSTS: SCOPE AND FUTURE. André Mortreux*, Olivier Coutelier Laboratoire de Catalyse de Lille UMR 8010 CNRS USTL-ENSCL, BP 90108, 59652 Villeneuve d’Ascq Cedex France Corresponding author.Tel +33320434993 ;Fax+33320434486 E-mail adress: [email protected] ABSTRACT This paper presents the evolution of alkyne metathesis since the early discoveries, essentially from the catalyst point of view. It is shown that although well defined carbynes may be useful for this reaction, further work has been made, aimed at the synthesis of new catalysts or catalytic systems, based on molybdenum precursors , associated or not with phenolic co- catalysts. The major objectives have been to obtain more functional groups tolerants catalysts, for their application in organic synthesis, including RCM for further stereoselective hydrogenation of the triple bond in the cycle, as well as for polymerization of aromatic diynes. -

Physical Sciences Content
Foreword In order to improve learning outcomes the Department of Basic Education conducted research to determine the specific areas that learners struggle with in Grade 12 examinations. The research included a trend analysis by subject experts of learner performance over a period of five years as well as learner examination scripts in order to diagnose deficiencies or misconceptions in particular content areas. In addition, expert teachers were interviewed to determine the best practicesto ensure mastery of thetopic by learners and improve outcomes in terms of quality and quantity. The results of the research formed the foundation and guiding principles for the development of the booklets. In each identified subject, key content areas were identified for the development of material that will significantly improve learner's conceptual understanding whilst leading to improved performance in the subject. The booklets are developed as part of a series of booklets, with each bookletfocussing onlyon one specific challenging topic. The selected content is explained in detail and include relevant concepts from Grades 10 - 12 to ensure conceptual understanding. The main purpose of these booklets is to assist learners to master the content starting from a basic conceptual level of understanding to the more advanced level. The content in each booklets is presented in an easy to understand manner including the use of mind maps, summaries and exercises to support understanding and conceptual progression. These booklets should ideally be used as part of a focussed revision or enrichment program by learners after the topics have been taught in class. The booklets encourage learners to take ownership of their own learning and focus on developing and mastery critical content and skills such as reading and higher order thinking skills. -

Heuristics for Searching Chemical Structures a Thesis Presented To
Heuristics for Searching Chemical Structures A Thesis presented to the Faculty of the Graduate School University of Missouri-Columbia In Partial Fulfillment of the Requirements for the Degree Master of Science by Nandini Basu Dr. Toni Kazic, Thesis Supervisor May 2007 The undersigned, appointed by the Dean of the Graduate School, have examined the thesis entitled HEURISTICS FOR SEARCHING CHEMICAL STRUCTURES presented by Nandini Basu A candidate for the degree of Master of Science And hereby certify that in their opinion it is worthy of acceptance. Dr. Toni Kazic Dr. Chi-Ren Shyu Dr. Mary Schaeffer ACKNOWLEDGMENTS I am deeply indebted to my research advisor Dr. Toni Kazic for giving me an opportunity to resume my research in and complete my Computer Science MS program. Without her guidance, support and encouragement this would not have been possible. I would like to thank Dr. Mary Polaco and Dr. Shyu for serving in my committee and helping me in my thesis. I would like to thank the Klotho team for helping me with structures. And also the other team members Amith, Archana, Avanthi, Bill, Deepthi, Gaurav, Raman, Jiahui, Fatten, Meeta and Michael. My friends made a difficult journey lot of fun and much easier, especially Cathy, Charu, Deepak, Karan, Karthik, Paco, Nikunj, Raj, Shiva, Shraddha, Sundeep and Vibha. All my relatives gave me constant encouragement and I would like to take this opportunity to thank them. And above all, I would like to thank my father Prabhat Kumar Basu, my mother Minoti Basu, my brother Pradipta Kumar Basu and my sister-in-law Sarmistha Basu. -

1 Pesticide Analysis by Mass Spectrometry Purpose
Pesticide Analysis by Mass Spectrometry Purpose: The purpose of this assignment is to introduce concepts of mass spectrometry (MS) as they pertain to the qualitative and quantitative analysis of organochlorine pesticides found in water samples. Learning Outcomes At the end of this assignment, students will be able to: 1. Explain the processes involved in the ionization of compounds in GCMS. 2. Predict isotopic distributions and identify chlorinated compounds by their MS isotopic signature and fragmentation patterns. 3. Identify and use unique ions for MS quantitation in complex samples. 4. Using a sampling scheme for Lake Nakuru, determine the concentrations of DDT from GCMS results and draw some conclusions as to whether the levels of DDT detected in Lake Nakuru water play a role in flamingos’ death. In the previous modules you have been introduced to the problem of flamingo die off events in Lake Nakuru, and the analytical chemistry considerations needed to address this problem including sampling, sample preparation, and gas chromatography. In addition to this discussion of MS analysis, the following module on validation of the analytical method addresses issues related to ensuring that the results obtained are of sufficient quality that they can be used to draw appropriate scientific conclusions. In this section, aspects of ionization and mass analysis are explored and example GCMS results are presented. Introduction This section is designed to provide an introduction to mass spectrometry (MS) in the context of its utilization as a gas chromatography detector. The basic concepts of ionization, mass analysis, isotope ratios, and mass spectral interpretation are introduced. -

Metallacycle–Based Nickel–Catalyzed Reductive Couplings and Reductive Cross–Electrophile Couplings
Metallacycle–Based Nickel–Catalyzed Reductive Couplings and Reductive Cross–Electrophile Couplings by Amie Renee Frank A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Chemistry) in the University of Michigan 2020 Doctoral Committee: Professor John Montgomery, Chair Associate Professor Amanda L. Garner Associate Professor Corinna S. Schindler Professor John P. Wolfe Amie Renee Frank [email protected] ORCID iD: 0000-0002-6155-3632 © Amie Renee Frank 2020 Dedication To my husband, Caleb Frank. Your love, patience, support and encouragement throughout this adventure makes me want to grow old with you even more. On to the next adventure. ii Acknowledgements I Would like to thank my advisor, Professor John Montgomery, for allowing me the opportunity to Work in his lab the past five years. His guidance and mentorship provided me With the tools necessary to grow as a scientist and develop as a person. Thank you for your support and the freedom to explore my own interests. I Would also like to thank Professor John Wolfe for allowing me the opportunity to rotate in his lab as a first–year graduate student. His support, advice and feedback throughout my graduate career Were alWays greatly appreciated. To the other members on my committee, Professor Corinna Schindler and Amanda Garner, thank you for being inspiring female role models in science. Their support, advice and feedback Were also extremely helpful throughout my graduate career. Additionally, I Would like to thank Professor Anne McNeil for her support as my original research proposal mentor. Lastly, I Would like to thank Professor Brian Coppola and Professor Adam Matzger for their support and advice during times of adversity. -

And Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes † † Kristian E
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. Review Cite This: Chem. Rev. 2019, 119, 2752−2875 pubs.acs.org/CR Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes † † Kristian E. Dalle, Julien Warnan, Jane J. Leung, Bertrand Reuillard, Isabell S. Karmel, and Erwin Reisner* Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, United Kingdom ABSTRACT: The synthesis of renewable fuels from abundant water or the greenhouse gas CO2 is a major step toward creating sustainable and scalable energy storage technologies. In the last few decades, much attention has focused on the development of nonprecious metal-based catalysts and, in more recent years, their integration in solid-state support materials and devices that operate in water. This review surveys the literature on 3d metal-based molecular catalysts and focuses on their immobilization on heterogeneous solid-state supports for electro-, photo-, and photoelectrocatalytic synthesis of fuels in aqueous media. The first sections highlight benchmark homogeneous systems using proton and CO2 reducing 3d transition metal catalysts as well as commonly employed methods for catalyst immobilization, including a discussion of supporting materials and anchoring groups. The subsequent sections elaborate on productive associations between molecular catalysts and a wide range of substrates based on carbon, quantum dots, metal oxide surfaces, and semiconductors. The molecule−material hybrid systems are organized as “dark” cathodes, colloidal photocatalysts, and photocathodes, and their figures of merit are discussed alongside system stability and catalyst integrity. -

Olefin Metathesis
Tetrahedron 60 (2004) 7117–7140 Olefin metathesis Robert H. Grubbs* The Arnold and Mabel Beckman Laboratory of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA Received 10 May 2004; accepted 11 May 2004 Abstract—Olefin metathesis has become a tool for synthetic organic and polymer chemists. Well-defined, functional group tolerant catalysts have allowed these advances. A discussion of the evolution of mechanistic understanding and early catalyst developments is followed by a description of recent advances in ruthenium based olefin metathesis catalysts. Catalysts improvements have led to new applications in ring closing metathesis, cross metathesis and materials synthesis. q 2004 Published by Elsevier Ltd. As with most catalytic processes, olefin metathesis was Chauvin proposed a new mechanism to explain a surprising found by accident. It was discovered as an outgrowth of the set of observations.7 He observed that in some cases where a study of Ziegler polymerizations with alternate metal pair-wise mechanism such as the ‘quasicyclobutane’ systems.1 By the late 60’s, the Phillips group developed a mechanism, predicted only the two olefins resulting from commercial process—the triolefin process—and made the pair-wise exchange of the two ends of the stating olefins, the scientific community aware of this unique reaction.2 My olefins resulting from cross products were observed very introduction to olefin metathesis occurred during a group early in the reaction. Although some assumptions would meeting while I was a postdoctoral fellow in Jim Collman’s allow the pair-wise mechanism to account for this result, group at Stanford.