Enantioselective Alkylation of Aldehydes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Photoredox Catalysis Photoredox Catalysis: a Mild, Operationally Simple Approach to the Synthesis of A-Trifluoromethyl Carbonyl Compounds Phong V
DOI: 10.1002/anie.201101861 Photoredox Catalysis Photoredox Catalysis: A Mild, Operationally Simple Approach to the Synthesis of a-Trifluoromethyl Carbonyl Compounds Phong V. Pham, David A. Nagib, and David W. C. MacMillan* The unique physical and chemical advantages conferred by the CÀF bond have led to the broad exploitation of this motif throughout the pharmaceutical,[1] materials,[2] and agrochem- ical[3] sectors. In drug design, for instance, incorporation of polyfluorinated alkyl groups, such as CF3 moieties, can profoundly impact the activity, metabolic stability, lipophilic- ity, and bioavailability of lead compounds.[1,4] Not surpris- ingly, the development of methods for the production of carbonyl-based synthons bearing a-CF3 substitution has emerged as a central objective in the field of chemical synthesis. Although important recent advances have been made toward this goal, there are currently few operationally simple methods for the conversion of enolates (or enolate equivalents) to a-trifluoromethylated carbonyl motifs. Stan- dard alkylation methods are generally not productive, due to the negative polarization of the trifluoromethyl moiety, thus specially tailored reagents have been developed to furnish an trifluoromethylation of a range of enolates or enolate [5] electrophilic CF3 equivalent. Alternatively, in recent years, a equivalents [Eq. (1)]. In this context, we elected to employ set of radical (Et3B/O2) and organometallic (Rh-catalyzed) enolsilanes and silylketene acetals as suitable enolic sub- approaches have been -

Electrochemistry and Photoredox Catalysis: a Comparative Evaluation in Organic Synthesis
molecules Review Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis Rik H. Verschueren and Wim M. De Borggraeve * Department of Chemistry, Molecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, box 2404, 3001 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-7693 Received: 30 March 2019; Accepted: 23 May 2019; Published: 5 June 2019 Abstract: This review provides an overview of synthetic transformations that have been performed by both electro- and photoredox catalysis. Both toolboxes are evaluated and compared in their ability to enable said transformations. Analogies and distinctions are formulated to obtain a better understanding in both research areas. This knowledge can be used to conceptualize new methodological strategies for either of both approaches starting from the other. It was attempted to extract key components that can be used as guidelines to refine, complement and innovate these two disciplines of organic synthesis. Keywords: electrosynthesis; electrocatalysis; photocatalysis; photochemistry; electron transfer; redox catalysis; radical chemistry; organic synthesis; green chemistry 1. Introduction Both electrochemistry as well as photoredox catalysis have gone through a recent renaissance, bringing forth a whole range of both improved and new transformations previously thought impossible. In their growth, inspiration was found in older established radical chemistry, as well as from cross-pollination between the two toolboxes. In scientific discussion, photoredox catalysis and electrochemistry are often mentioned alongside each other. Nonetheless, no review has attempted a comparative evaluation of both fields in organic synthesis. Both research areas use electrons as reagents to generate open-shell radical intermediates. Because of the similar modes of action, many transformations have been translated from electrochemical to photoredox methodology and vice versa. -

A Synergistic LUMO Lowering Strategy Using Lewis Acid Catalysis in Water to Enable Photoredox Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue A synergistic LUMO lowering strategy using Lewis acid catalysis in water to enable photoredox Cite this: Chem. Sci.,2018,9,7096 – All publication charges for this article catalytic, functionalizing C C cross-coupling of have been paid for by the Royal Society † of Chemistry styrenes Elisabeth Speckmeier, Patrick J. W. Fuchs and Kirsten Zeitler * Easily available a-carbonyl acetates serve as convenient alkyl radical source for an efficient, photocatalytic cross-coupling with a great variety of styrenes. Activation of electronically different a-acetylated acetophenone derivatives could be effected via LUMO lowering catalysis using a superior, synergistic combination of water and (water-compatible) Lewis acids. Deliberate application of fac-Ir(ppy)3 as photocatalyst to enforce an oxidative quenching cycle is crucial to the success of this (umpolung type) transformation. Mechanistic particulars of this dual catalytic coupling reaction have been studied in detail using both Stern–Volmer and cyclic voltammetry Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Received 12th May 2018 experiments. As demonstrated in more than 30 examples, our water-assisted LA/photoredox Accepted 17th July 2018 catalytic activation strategy allows for excess-free, equimolar radical cross-coupling and subsequent DOI: 10.1039/c8sc02106f formal Markovnikov hydroxylation to versatile 1,4-difunctionalized products in good to excellent rsc.li/chemical-science yields. redox properties: with an oxidation potential of only 0.66 V vs. Introduction À SCE (Eox(Brc/Br ) ¼ 0.66 V) mesolytically cleaved bromide Visible light photocatalysis as a powerful tool for the selective anions can easily get oxidized to the corresponding bromine This article is licensed under a generation of radicals has given rise to numerous unprece- radicals and hence potentially may also form elemental Br2 by 8–10 dented C–C-coupling reactions in recent years.1 Especially alkyl most common photocatalysts. -

Merging Photoredox Catalysis with Organocatalysis
REPORTS 8. C. Resini, T. Montanari, G. Busca, J.-M. Jehng, 17. M. Salmeron, R. Schlögl, Surf. Sci. Rep. 63, 169 (2008). für Synchrotronstrahlung for support of in situ XPS I. E. Wachs, Catal. Today 99, 105 (2005). 18. T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, measurements; and M. A Smith for helpful discussions. 9. T. G. Alkhazov, E. A. Ismailov, A. Yu, A. I. Kozharov, J. McLaughlin, N. M. D. Brown, Carbon 43, 153 (2005). Supported by the CANAPE project of the 6th Framework Kinet. Katal. 19, 611 (1978). 19. L. M. Madeira, M. F. Portela, Catal. Rev. 44, 247 (2002). Programme of European Commission and the ENERCHEM 10. M. S. Kane, L. C. Kao, R. K. Mariwala, D. F. Hilscher, 20. F. Atamny et al., Mol. Phys. 76, 851 (1992). project of the Max Planck Society. H. C. Foley, Ind. Eng. Chem. Res. 35, 3319 (1996). 21. M. L. Toebes, J. M. P. van Heeswijk, J. H. Bitter, 11. M. F. R. Pereira, J. J. M. Órfão, J. L. Figueiredo, A. J. van Dillen, K. P. de Jong, Carbon 42, 307 (2004). Appl. Catal. A 218, 307 (2001). 22. A. M. Puziy, O. I. Poddubnaya, A. M. Ziatdinov, Appl. Surf. Supporting Online Material www.sciencemag.org/cgi/content/full/322/5898/73/DC1 12. J. Zhang et al., Angew. Chem. Int. Ed. 46, 7319 (2007). Sci. 252, 8036 (2006). Materials and Methods 13. G. Mestl, N. I. Maksimova, N. Keller, V. V. Roddatis, 23. S. J. Clark et al., Z. Kristallogr. 220, 567 (2005). R. Schlögl, Angew. -

Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis Christopher K
Review pubs.acs.org/CR Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis Christopher K. Prier, Danica A. Rankic, and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States References 5360 1. INTRODUCTION CONTENTS A fundamental aim in the field of catalysis is the development 1. Introduction 5322 of new modes of small molecule activation. One approach 2+ toward the catalytic activation of organic molecules that has 2. Photochemistry of Ru(bpy)3 5323 3. Net Reductive Reactions 5324 received much attention recently is visible light photoredox 3.1. Reduction of Electron-Poor Olefins 5324 catalysis. In a general sense, this approach relies on the ability of 3.2. Reductive Dehalogenation 5326 metal complexes and organic dyes to engage in single-electron- 3.3. Reductive Cleavage of Sulfonium and transfer (SET) processes with organic substrates upon Sulfonyl Groups 5328 photoexcitation with visible light. 3.4. Nitrogen Functional Group Reductions 5329 Many of the most commonly employed visible light 3.5. Radical Cyclizations 5330 photocatalysts are polypyridyl complexes of ruthenium and iridium, and are typified by the complex tris(2,2′-bipyridine) 3.6. Reductive Epoxide and Aziridine Opening 5331 2+ 3.7. Reduction-Labile Protecting Groups 5331 ruthenium(II), or Ru(bpy)3 (Figure 1). These complexes 4. Net Oxidative Reactions 5332 4.1. Functional Group Oxidations 5332 4.2. Oxidative Removal of the PMB Group 5334 4.3. Oxidative Biaryl Coupling 5335 4.4. Oxidative Generation of Iminium Ions 5335 4.5. Azomethine Ylide [3 + 2] Cycloadditions 5338 4.6. -

Difunctionalization of Alkenes and Alkynes Via Intermolecular Radical and Nucleophilic Additions
molecules Review Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions Hongjun Yao 1,†, Wenfei Hu 2,† and Wei Zhang 2,* 1 College of Biological Science and Technology, Beijing Forestry University, 35 Qinghua East Road, Beijing 100083, China; [email protected] 2 Department of Chemistry, University of Massachusetts Boston, 100 Morrissey Boulevard, Boston, MA 02125, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-617-287-6147 † These authors contributed equally to this work. Abstract: Popular and readily available alkenes and alkynes are good substrates for the preparation of functionalized molecules through radical and/or ionic addition reactions. Difunctionalization is a topic of current interest due to its high efficiency, substrate versatility, and operational simplicity. Presented in this article are radical addition followed by oxidation and nucleophilic addition reactions for difunctionalization of alkenes or alkynes. The difunctionalization could be accomplished through 1,2-addition (vicinal) and 1,n-addition (distal or remote) if H-atom or group-transfer is involved in the reaction process. A wide range of moieties, such as alkyl (R), perfluoroalkyl (Rf), aryl (Ar), hydroxy (OH), alkoxy (OR), acetatic (O2CR), halogenic (X), amino (NR2), azido (N3), cyano (CN), as well as sulfur- and phosphorous-containing groups can be incorporated through the difunctional- ization reactions. Radicals generated from peroxides or single electron transfer (SET) agents, under photoredox or electrochemical reactions are employed for the reactions. Keywords: difunctionalization; radical; nucleophilic; alkene; alkyne; photoredox; single electron transfer; vicinal-difunctionalization; distal-difunctionalization; photoredox; electrochemical reaction Citation: Yao, H.; Hu, W.; Zhang, W. Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions. -

Photoredox Catalysis in Organic Chemistry Megan H
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Perspective pubs.acs.org/joc Photoredox Catalysis in Organic Chemistry Megan H. Shaw, Jack Twilton, and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States ABSTRACT: In recent years, photoredox catalysis has come to the forefront in organic chemistry as a powerful strategy for the activation of small molecules. In a general sense, these approaches rely on the ability of metal complexes and organic dyes to convert visible light into chemical energy by engaging in single-electron transfer with organic substrates, thereby generating reactive intermediates. In this Perspective, we highlight the unique ability of photoredox catalysis to expedite the development of completely new reaction mechanisms, with particular emphasis placed on multicatalytic strategies that enable the construction of challenging carbon−carbon and carbon− heteroatom bonds. ■ INTRODUCTION this catalytic platform to organic synthesis begun to be fully Over the last century, the discovery, development, and use of realized. A key factor in the recent yet rapid growth of this activation platform has been the recognition that readily light-mediated catalysis has enabled the invention of a wide accessible metal polypyridyl complexes and organic dyes can variety of nontraditional bond constructions in organic facilitate the conversion -

COMMUNICATION Asymmetric Organocatalysis and Photoredox
Asymmetric Organocatalysis and Photoredox Catalysis for the α-Functionalization of Tetrahydroisoquinolines Item Type Article Authors Hou, Hong; Zhu, Shaoqun; Atodiresei, Iuliana; Rueping, Magnus Citation Hou H, Zhu S, Atodiresei I, Rueping M (2018) Asymmetric Organocatalysis and Photoredox Catalysis for the α- Functionalization of Tetrahydroisoquinolines. European Journal of Organic Chemistry 2018: 1277–1280. Available: http:// dx.doi.org/10.1002/ejoc.201800117. Eprint version Post-print DOI 10.1002/ejoc.201800117 Publisher Wiley Journal European Journal of Organic Chemistry Rights This is the peer reviewed version of the following article: Asymmetric Organocatalysis and Photoredox Catalysis for the α-Functionalization of Tetrahydroisoquinolines, which has been published in final form at http://doi.org/10.1002/ejoc.201800117. This article may be used for non-commercial purposes in accordance With Wiley Terms and Conditions for self-archiving. Download date 29/09/2021 05:30:43 Link to Item http://hdl.handle.net/10754/627347 COMMUNICATION Asymmetric Organocatalysis and Photoredox Catalysis for the α- Functionalization of Tetrahydroisoquinolines Hong Hou,[a,b] Shaoqun Zhu,[a] Iuliana Atodiresei[a] and Magnus Rueping[a,c]* Abstract: The asymmetric α-alkylation of tetrahydroisoquinolines with particular, cyclic ketones were selected which are more cyclic ketones has been accomplished in the presence of a combined challenging substrates in the enantioselective oxidative coupling catalytic system consisting of a visible-light photoredox catalyst and a reaction with tertiary amines (Scheme 1).[12] chiral primary amine organocatalyst. The desired products were obtained in good yields, high enantioselectivity and good to excellent diastereoselectivity. O + hv, Photocatalyst Ar N * Ar Organocatalyst O * The harnessing of visible light sensitization as a powerful strategy for initiating organic reactions is attractive and becoming an important tool in synthesis due to its favorable features, including low cost, convenience and environmentally Scheme 1. -

Advances in Visible-Light-Mediated Carbonylative Reactions Via Carbon Monoxide (CO) Incorporation
catalysts Review Advances in Visible-Light-Mediated Carbonylative Reactions via Carbon Monoxide (CO) Incorporation Vinayak Botla 1, Aleksandr Voronov 1, Elena Motti 1, Carla Carfagna 2, Raffaella Mancuso 3 , Bartolo Gabriele 3 and Nicola Della Ca’ 1,* 1 Department of Chemistry, Life Sciences and Environmental Sustainability (SCVSA), University of Parma, Parco Area delle Scienze, 17/A, 43124 Parma, Italy; [email protected] (V.B.); [email protected] (A.V.); [email protected] (E.M.) 2 Department of Industrial Chemistry “T. Montanari”, University of Bologna, 40136 Bologna, Italy; [email protected] 3 Laboratory of Industrial and Synthetic Organic Chemistry (LISOC), Department of Chemistry and Chemical Technologies, University of Calabria, Via P. Bucci 12/C, 87036 Arcavacata di Rende Cosenza, Italy; [email protected] (R.M.); [email protected] (B.G.) * Correspondence: [email protected]; Tel.:+39-0521-905676 Abstract: The abundant and inexpensive carbon monoxide (CO) is widely exploited as a C1 source for the synthesis of both fine and bulk chemicals. In this context, photochemical carbonylation reactions have emerged as a powerful tool for the sustainable synthesis of carbonyl-containing compounds (esters, amides, ketones, etc.). This review aims at giving a general overview on visible light-promoted carbonylation reactions in the presence of metal (Palladium, Iridium, Cobalt, Ruthenium, Copper) and organocatalysts as well, highlighting the main features of the presented protocols and providing Citation: Botla, V.; Voronov, A.; useful insights on the reaction mechanisms. Motti, E.; Carfagna, C.; Mancuso, R.; Gabriele, B.; Della Ca’, N. Advances Keywords: carbon monoxide; carbonylation; photochemical reactions; visible light; carbonyl- in Visible-Light-Mediated containing compounds Carbonylative Reactions via Carbon Monoxide (CO) Incorporation. -

Photoredox-Catalyzed Oxo-Amination of Aryl Cyclopropanes
ARTICLE https://doi.org/10.1038/s41467-019-12403-2 OPEN Photoredox-catalyzed oxo-amination of aryl cyclopropanes Liang Ge1, Ding-Xing Wang1, Renyi Xing1,DiMa1, Patrick J. Walsh 2* & Chao Feng 1* Cyclopropanes represent a class of versatile building blocks in modern organic synthesis. While the release of ring strain offers a thermodynamic driving force, the control of selectivity for C–C bond cleavage and the subsequent regiochemistry of the functionalization remains fi 1234567890():,; dif cult, especially for unactivated cyclopropanes. Here we report a photoredox-coupled ring- opening oxo-amination of electronically unbiased cyclopropanes, which enables the expe- dient construction of a host of structurally diverse β-amino ketone derivatives. Through one electron oxidation, the relatively inert aryl cyclopropanes are readily converted into reactive radical cation intermediates, which in turn participate in the ensuing ring-opening functio- nalizations. Based on mechanistic studies, the present oxo-amination is proposed to proceed through an SN2-like nucleophilic attack/ring-opening manifold. This protocol features wide substrate scope, mild reaction conditions, and use of dioxygen as an oxidant both for catalyst regeneration and oxygen-incorporation. Moreover, a one-pot formal aminoacylation of olefins is described through a sequential cyclopropanation/oxo-amination. 1 Institute of Advanced Synthesis (IAS), School of Chemistry and Molecular Engineering, Nanjing Tech University, 30 South Puzhu Road, 211816 Nanjing,P.R. China. 2 Roy and -

A Combination of Visible‐Light Organophotoredox Catalysis And
A Combination of Visible‐Light Organophotoredox Catalysis and Asym‐ metric Organocatalysis for the Enantioselective Mannich Reaction of Dihydroquinoxalinones with Ketones. Jaume Rostoll-Berenguer,a Gonzalo Blay,a M. Carmen Muñoz,b José R. Pedro,a* Carlos Vilaa* a Departament de Química Orgànica, Facultat de Química, Universitat de València, Dr. Moliner 50, 46100 Burjassot, Valèn- cia (Spain). E-mail: [email protected], [email protected], b Departament de Física Aplicada, Universitat Politècnica de València, Camino de Vera s/n, 46022 València (Spain). Supporting Information Placeholder ABSTRACT: An enantioselective photooxidative Mannich reaction of dihydroquinoxalinones with ketones by the merger of organ- ophotoredox and asymmetric organocatalysis is described. This protocol features a very mild reaction conditions using simple and cheap catalysts (Eosin Y and (S)-Proline) for the synthesis of chiral quinoxaline derivatives with good to high yields (up to 94%) and excellent enantioselectivities (up to 99% ee). The employment of visible-light photoredox catalysis have The enantioselective synthesis of chiral amines is, generally, become a hot topic in synthetic chemistry in the last decade, due a remarkable research area owing to the importance of chiral to its significant advantages such as low cost, safe, renewable, nitrogen‐containing compounds for medicinal, pharmaceutical abundant, and environmentally friendliness. 1,2 On the other and agrochemical chemistry.8 In this context, since the pioneer- hand, control of chirality is one -
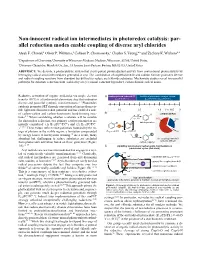
Non-Innocent Radical Ion Intermediates in Photoredox Catalysis: Par- Allel Reduction Modes Enable Coupling of Diverse Aryl Chlorides Alyah F
Non-innocent radical ion intermediates in photoredox catalysis: par- allel reduction modes enable coupling of diverse aryl chlorides Alyah F. Chmiel,1 Oliver P. Williams,1 Colleen P. Chernowsky,1 Charles S. Yeung,*,2 and Zachary K. Wickens*,1 1Department of Chemistry, University of Wisconsin-Madison; Madison, Wisconsin, 53706, United States. 2Discovery Chemistry, Merck & Co., Inc., 33 Avenue Louis Pasteur, Boston, MA 02115, United States ABSTRACT: We describe a photocatalytic system that elicits potent photoreductant activity from conventional photocatalysts by leveraging radical anion intermediates generated in situ. The combination of isophthalonitrile and sodium formate promotes diverse aryl radical coupling reactions from abundant but difficult to reduce aryl chloride substrates. Mechanistic studies reveal two parallel pathways for substrate reduction both enabled by a key terminal reductant byproduct, carbon dioxide radical anion. Reductive activation of organic molecules via single electron A underexplored reduced PC traditional photoredox catalyst criteria: •– + •- transfer (SET) is a fundamental elementary step that underpins E1/2(PC/PC *) E1/2(PC /PC*, E1/2(PC/PC ) diverse and powerful synthetic transformations.1–4 Photoredox catalysis promotes SET through conversion of energy from vis- ible light into chemical redox potential and has enabled a suite –3.0 –2.0 –1.0 V vs SCE 0 B of carbon-carbon and carbon-heteroatom bond-forming reac- – + H commercial ArX 5–9 e H tions. When considering whether a substate will be suitable or H ArCl for photoredox reduction, two primary catalyst parameters are ArBr •+ •– Cl dehalogenation initially considered: (1) E1/2(PC /PC*) and (2) E1/2(PC/PC – ).7,10,11 These values reflect redox potentials bounded by the en- e ergy of photons in the visible region, a limitation compounded Ered = –2.8 V Ered = +0.05 V ArI 12 by energy losses to intersystem crossing.