Consumer Medicine Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effects of Pitavastatin, Atorvastatin, and Rosuvastatin on the Risk Of
biomedicines Article Effects of Pitavastatin, Atorvastatin, and Rosuvastatin on the Risk of New-Onset Diabetes Mellitus: A Single-Center Cohort Study Wei-Ting Liu 1, Chin Lin 2,3,4, Min-Chien Tsai 5, Cheng-Chung Cheng 6, Sy-Jou Chen 7,8, Jun-Ting Liou 6 , Wei-Shiang Lin 6, Shu-Meng Cheng 6, Chin-Sheng Lin 6,* and Tien-Ping Tsao 6,9,* 1 Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 2 School of Public Health, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 3 School of Medicine, National Defense Medical Center, Taipei 11490, Taiwan 4 Graduate Institute of Life Sciences, National Defense Medical Center, Taipei 11490, Taiwan, 5 Department of Physiology and Biophysics, Graduate Institute of Physiology, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 6 Division of Cardiology, Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] (C.-C.C.); [email protected] (J.-T.L.); [email protected] (W.-S.L.); [email protected] (S.-M.C.) 7 Department of Emergency Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 8 Graduate Institute of Injury Prevention and Control, College of Public Health and Nutrition, Taipei Medical University, Taipei 11031, Taiwan 9 Division of Cardiology, Cheng Hsin General Hospital, Taipei 11220, Taiwan * Correspondence: [email protected] (C.-S.L.); [email protected] (T.-P.T.); Tel.: +886-2-6601-2656 (C.-S.L.); +886-2-2826-4400 (T.-P.T.) Received: 25 October 2020; Accepted: 11 November 2020; Published: 13 November 2020 Abstract: Statins constitute the mainstay treatment for atherosclerotic cardiovascular disease, which is associated with the risk of new-onset diabetes mellitus (NODM). -

Deliverable 5.A Interim Report on the Study Results APPENDIX 2
Deliverable 5.a Interim report on the study results APPENDIX 2: Algorithms used to identify study variables for service contract EMA/2011/38/CN ‐ PIOGLITAZONE November 28th 2012 D5.a Interim report on the study results (Appendix 2) for Service Contract EMA/2011/38/CN PIOGLITAZONE Author(s): Vera Ehrenstein (AUH‐AS) APPENDIX 2. ALGORITHMS USED TO IDENTIFY STUDY VARIABLES Algorithms for AU Database DISEASE/CONDITION ICD-8 CODE (1977-1993) ICD-10 CODE (1994-) Diabetes type 2 250.00; 250.06; 250.07; 250.09 E11.0; E11.1; E11.9 Cancer of bladder 188 C67 Haematuria N/A R31 Haematuria, unspecified B18, K70.0–K70.3, K70.9, K71, K73, Mild hepatic impairment 571, 573.01, 573.04 K74, K76.0 Moderate to severe hepatic 070.00, 070.02, 070.04, 070.06, B15.0, B16.0, B16.2, B19.0, K70.4, impairment 070.08, 573.00, 456.00–456.09 K72, K76.6, I85 Acute myocardial infarction 410 I21-I23 Acute coronary syndrome 410, 413 I20-I24 Ischemic heart disease 410-414 I20-I25 427.09, 427.10, 427.11, 427.19, Congestive heart failure I50, I11.0, I13.0,I13.2 428.99, 782.49; Acute renal failure N/A N17 Diabetic coma N/A E10.0, E11.0, E12.0,E13.0, E14.0 Diabetic acidosis N/A E10.1, E11.1, E12.1,E13.1, E14.1 F10.1-F10.9, G31.2, G62.1, G72.1, Alcoholism 291, 303, 577.10, 571.09, 571.10 I42.6, K29.2, K86.0, Z72.1 Obesity 277.99 E65-E66 D5.a Interim report on the study results (Appendix 2) for Service Contract EMA/2011/38/CN PIOGLITAZONE Author(s): Vera Ehrenstein (AUH‐AS) Algorithms for defining acute events in Denmark, ICD-10 code Event ICD-10 code I21.x, I23.x http://apps.who.int/classifications/icd10/browse/2010/en#/I21 -

Zetia® (Ezetimibe) Tablets
29480958T REV 14 ZETIA® (EZETIMIBE) TABLETS DESCRIPTION ZETIA (ezetimibe) is in a class of lipid-lowering compounds that selectively inhibits the intestinal absorption of cholesterol and related phytosterols. The chemical name of ezetimibe is 1-(4-fluorophenyl)- 3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone. The empirical formula is C24H21F2NO3. Its molecular weight is 409.4 and its structural formula is: OH OH S SR N F F O Ezetimibe is a white, crystalline powder that is freely to very soluble in ethanol, methanol, and acetone and practically insoluble in water. Ezetimibe has a melting point of about 163°C and is stable at ambient temperature. ZETIA is available as a tablet for oral administration containing 10 mg of ezetimibe and the following inactive ingredients: croscarmellose sodium NF, lactose monohydrate NF, magnesium stearate NF, microcrystalline cellulose NF, povidone USP, and sodium lauryl sulfate NF. CLINICAL PHARMACOLOGY Background Clinical studies have demonstrated that elevated levels of total cholesterol (total-C), low density lipoprotein cholesterol (LDL-C) and apolipoprotein B (Apo B), the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of high density lipoprotein cholesterol (HDL-C) are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including very-low- density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis. The independent effect of raising HDL-C or lowering triglycerides (TG) on the risk of coronary and cardiovascular morbidity and mortality has not been determined. -

Bempedoic Acid) Tablets, for Oral Use Most Common (Incidence ≥ 2% and Greater Than Placebo) Adverse Reactions Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION • Tendon Rupture: Tendon rupture has occurred. Discontinue NEXLETOL These highlights do not include all the information needed to use at the first sign of tendon rupture. Avoid NEXLETOL in patients who NEXLETOL™ safely and effectively. See full prescribing information have a history of tendon disorders or tendon rupture. (5.2) for NEXLETOL. --------------------------------ADVERSE REACTIONS---------------------------- NEXLETOL (bempedoic acid) tablets, for oral use Most common (incidence ≥ 2% and greater than placebo) adverse reactions Initial U.S. Approval: 2020 are upper respiratory tract infection, muscle spasms, hyperuricemia, back pain, abdominal pain or discomfort, bronchitis, pain in extremity, anemia, ----------------------------INDICATIONS AND USAGE-------------------------- and elevated liver enzymes. (6.1) NEXLETOL is an adenosine triphosphate-citrate lyase (ACL) inhibitor indicated as an adjunct to diet and maximally tolerated statin therapy for the To report SUSPECTED ADVERSE REACTIONS, contact Esperion at treatment of adults with heterozygous familial hypercholesterolemia or 833-377-7633 (833 ESPRMED) or FDA at 1-800-FDA-1088 or established atherosclerotic cardiovascular disease who require additional www.fda.gov/medwatch. lowering of LDL-C. (1) --------------------------------DRUG INTERACTIONS---------------------------- Limitations of Use: The effect of NEXLETOL on cardiovascular morbidity • Simvastatin: Avoid concomitant use of NEXLETOL with simvastatin and mortality has not been -

PCSK9 Inhibition in Familial Hypercholesterolemia: a Revolution in Treatment
PCSK9 inhibition in familial hypercholesterolemia: A revolution in treatment Frederick Raal FCP(SA), FRCP, FCRPC, Cert Endo, MMED, PHD Head, Division of Endocrinology & Metabolism Director, Carbohydrate and Lipid Metabolism Research Unit Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa A case of severe hypercholesterolemia Mrs. TDK 50 year old female. Well. Non smoker and no other risk factors for cardiovascular disease. Serum cholesterol measured because of positive family history of coronary artery disease, her father having undergone coronary bypass surgery at age 52. On examination she had arcus cornealis as well as marked thickening of her tendo-Achilles. Mrs TDK, a case of severe hypercholesterolemia Fasting lipid profile Total cholesterol: 15.6 mmol/L Triglycerides: 1.34 mmol/L HDL-cholesterol: 1.8 mmol/L LDL-cholesterol 13.2 mmol/L Heterozygous vs Homozygous FH Heterozygous FH Homozygous FH Prevalence 1:200- 500 Prevalence 1:300 000 -1000 000 LDL-cholesterol LDL-cholesterol 5.0-12 mmol/L 12-20 mmol/L (190-500mg/dL) (500–1000 mg/dL) CHD onset usually age 30-60 yrs CHD onset in early childhood Most patients respond to drug therapy, but individual response Poorly responsive to quite variable lipid-lowering drug therapy Nordestgaard B.G. et al. Eur Heart J 2013;34:3478-3490 Diagnostic definition of homozygous familial hypercholesterolemia . Genetic confirmation of 2 mutant alleles at the LDL receptor, APOB, PCSK9, or ARH adaptor protein gene locus OR . An untreated LDL cholesterol of 13 mmol/L (>500 mg/dL) or treated LDL cholesterol 7.8 mmol/L (≥300 mg/dL) or treated non-HDL cholesterol 8.5 mmol/L (≥330 mg/dL) together with either: - Cutaneous or tendonous xanthoma before age 10 years OR - Elevated LDL cholesterol levels before lipid-lowering therapy consistent with heterozygous FH in both parents* * Except in the case of ARH Raal FJ, Santos RD. -

Quasi-Experimental Health Policy Research: Evaluation of Universal Health Insurance and Methods for Comparative Effectiveness Research
Quasi-Experimental Health Policy Research: Evaluation of Universal Health Insurance and Methods for Comparative Effectiveness Research The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Garabedian, Laura Faden. 2013. Quasi-Experimental Health Policy Research: Evaluation of Universal Health Insurance and Methods for Comparative Effectiveness Research. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:11156786 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Quasi-Experimental Health Policy Research: Evaluation of Universal Health Insurance and Methods for Comparative Effectiveness Research A dissertation presented by Laura Faden Garabedian to The Committee on Higher Degrees in Health Policy in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Health Policy Harvard University Cambridge, Massachusetts March 2013 © 2013 – Laura Faden Garabedian All rights reserved. Professor Stephen Soumerai Laura Faden Garabedian Quasi-Experimental Health Policy Research: Evaluation of Universal Health Insurance and Methods for Comparative Effectiveness Research Abstract This dissertation consists of two empirical papers and one methods paper. The first two papers use quasi-experimental methods to evaluate the impact of universal health insurance reform in Massachusetts (MA) and Thailand and the third paper evaluates the validity of a quasi- experimental method used in comparative effectiveness research (CER). My first paper uses interrupted time series with data from IMS Health to evaluate the impact of Thailand’s universal health insurance and physician payment reform on utilization of medicines for three non-communicable diseases: cancer, cardiovascular disease and diabetes. -

Colesevelam Hydrochloride (Cholestagel) a New, Potent Bile Acid Sequestrant Associated with a Low Incidence of Gastrointestinal Side Effects
ORIGINAL INVESTIGATION Colesevelam Hydrochloride (Cholestagel) A New, Potent Bile Acid Sequestrant Associated With a Low Incidence of Gastrointestinal Side Effects Michael H. Davidson, MD; Maureen A. Dillon; Bruce Gordon, MD; Peter Jones, MD; Julie Samuels, MD; Stuart Weiss, MD; Jonathon Isaacsohn, MD; Phillip Toth, MD; Steven K. Burke, MD Objectives: To compare colesevelam hydrochloride mg/dL) (19.1%) in the 3.75-g/d colesevelam treatment (Cholestagel), a nonabsorbed hydrogel with bile acid– group. Low-density lipoprotein cholesterol concentra- sequestering properties, with placebo for its lipid- tions at the end of treatment were significantly reduced lowering efficacy, its effects on laboratory and clinical from baseline levels in the 3.0- and 3.75-g/d colesevelam safety parameters, and the incidence of adverse events. treatment groups (P = .01 and P,.001, respectively). To- tal cholesterol levels demonstrated a similar response to Methods: Following diet and placebo lead-in periods, colesevelam treatment, with an 8.1% decrease from base- placebo or colesevelam was administered at 4 dosages (1.5, line in the 3.75-g/d treatment group (P<.001). High- 2.25, 3.0, or 3.75 g/d) for 6 weeks with morning and density lipoprotein cholesterol levels rose significantly evening meals to men and women with hypercholester- in the 3.0- and 3.75-g/d colesevelam treatment groups, olemia (low-density lipoprotein cholesterol level .4.14 by 11.2% (P = .006) and 8.1% (P = .02), respectively. mmol/L [.160 mg/dL]). Patients returned to the clinic Median triglyceride levels did not change from baseline, every 2 weeks throughout the treatment period for lipid nor were there any significant differences between parameter measurements and adverse event assess- treatment groups. -

06. Barter Evolving Concepots PACE.Pdf
Evolving concepts in management of patients at increased CV risk Philip Barter, MD Sydney, Australia Evolving concepts in management of patients at increased CV risk? Philip Barter School of Medical Sciences University of New South Wales Sydney, Australia Disclosures Received honorariums for participating as a consultant or as a member of advisory boards for AMGEN, AstraZeneca, CSL-Behring, Lilly, Merck, Novartis, Pfizer and Sanofi and for giving lectures for AMGEN, AstraZeneca, Merck and Pfizer. Major risk factors for Atherosclerotic Cardiovascular Disease (ASCVD) • Age • Gender • Smoking • Elevated LDL-C • Elevated triglyceride-rich lipoproteins • Reduced HDL-C • Elevated blood pressure • Diabetes • Abdominal obesity Modifiable risk factors for ASCVD • Smoking • Elevated LDL-C • Elevated triglyceride-rich lipoproteins • Reduced HDL-C • Elevated blood pressure • Diabetes • Abdominal obesity Modifiable risk factors for ASCVD • Smoking • Elevated LDL-C • Elevated triglyceride-rich lipoproteins • Reduced HDL-C • Elevated blood pressure • Diabetes • Abdominal obesity Treatment with statins has been shown in many trials to reduce the risk of having an atherosclerotic cardiovascular event In these statin trials, the more the LDL-C is reduced, the greater is the reduction in risk of having an event. Relationship of CVD events to LDL-C reduction achieved in statin clinical trials CTT Collaboration. Lancet 2005; 366:1267-78; Lancet 2010;376:1670-81. And the lower the achieved level of LDL-C, the lower the risk of having an event Secondary Prevention -

CP.PMN.237 Bempedoic Acid (Nexletol)
Clinical Policy: Bempedoic Acid (Nexletol), Bempedoic Acid/Ezetimibe (Nexlizet) Reference Number: CP.PMN.237 Effective Date: 09.01.20 Last Review Date: 02.21 Revision Log Line of Business: Commercial, HIM, Medicaid See Important Reminder at the end of this policy for important regulatory and legal information. Description The following are adenosine triphosphate-citrate lyase (ACL) inhibitors requiring prior authorization: bempedoic acid (Nexletol™) and bempedoic acid/ezetimibe (Nexlizet™). Nexlizet contains ezetimibe, which is a cholesterol absorption inhibitor. FDA Approved Indication(s) Nexletol and Nexlizet are indicated for use as adjuncts to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia (HeFH) or established atherosclerotic cardiovascular disease (ASCVD) who require additional lowering of low-density lipoprotein cholesterol (LDL-C). Limitation(s) of use: The effect of Nexletol and Nexlizet on cardiovascular morbidity and mortality has not been determined. Policy/Criteria Provider must submit documentation (such as office chart notes, lab results or other clinical information) supporting that member has met all approval criteria. It is the policy of health plans affiliated with Centene Corporation® that Nexletol and Nexlizet are medically necessary when the following criteria are met: I. Initial Approval Criteria A. Heterozygous Familial Hypercholesterolemia and Atherosclerotic Cardiovascular Disease (must meet all): 1. Diagnosis of one of the following (a or b): a. ASCVD as evidenced by a history of any one of the following conditions (i-vii): i. Acute coronary syndromes; ii. Clinically significant coronary heart disease (CHD) diagnosed by invasive or noninvasive testing (such as coronary angiography, stress test using treadmill, stress echocardiography, or nuclear imaging); iii. -

Bempedoic Acid, Bempedoic Acid/Ezetimibe (Nexletol™, Nexlizet™) EOCCO POLICY Policy Type: PA/SP Pharmacy Coverage Policy: EOCCO182
bempedoic acid, bempedoic acid/ezetimibe (Nexletol™, Nexlizet™) EOCCO POLICY Policy Type: PA/SP Pharmacy Coverage Policy: EOCCO182 Description Bempedoic acid, bempedoic acid/ezetimibe (Nexletol, Nexlizet) is an orally administered adenosine triphosphate-citrate lyase inhibitor, and ezetimibe is an intestinal cholesterol absorption inhibitor . Length of Authorization Initial: six months Renewal: 12 months Quantity Limits Product Name Dosage Form Indication Quantity Limit As an adjunct to diet and bempedoic acid maximally tolerated statin 180 mg tablets 30 tablets/30 days (Nexletol) therapy for the treatment of adults with heterozygous familial hypercholesterolemia or bempedoic established atherosclerotic 180 mg/10 mg tablets acid/ezetimibe cardiovascular disease who 30 tablets/30 days (Nexlizet) require additional lowering of LDL-C Initial Evaluation I. Bempedoic acid, bempedoic acid/ezetimibe (Nexletol, Nexlizet) may be considered medically necessary when the following criteria below are met: A. Member is 18 years of age or older; AND B. Therapy is prescribed by, or in consultation with, a provider specializing in lipid management (e.g. cardiology, lipidology, endocrinology); AND C. Therapy with a maximally tolerated statin for at least an 8-week duration has been ineffective; AND 1. The member continues to have an LDL-cholesterol level greater than or equal to 70 mg/dL while on maximally tolerated statin therapy; AND 2. The member will continue maximally tolerated statin therapy in combination with bempedoic acid, bempedoic acid/ezetimibe (Nexletol, Nexlizet); AND 1 bempedoic acid, bempedoic acid/ezetimibe (Nexletol™, Nexlizet™) EOCCO POLICY 3. The member will not use bempedoic acid, bempedoic acid/ezetimibe (Nexletol, Nexlizet) in combination with simvastatin (Zocor) >20 mg or pravastatin (Pravachol) >40 mg; OR i. -

Lipid Lowering Drugs Prescription and the Risk of Peripheral Neuropathy
1047 J Epidemiol Community Health: first published as 10.1136/jech.2003.013409 on 16 November 2004. Downloaded from RESEARCH REPORT Lipid lowering drugs prescription and the risk of peripheral neuropathy: an exploratory case-control study using automated databases Giovanni Corrao, Antonella Zambon, Lorenza Bertu`, Edoardo Botteri, Olivia Leoni, Paolo Contiero ............................................................................................................................... J Epidemiol Community Health 2004;58:1047–1051. doi: 10.1136/jech.2003.013409 Study objective: Although lipid lowering drugs are effective in preventing morbidity and mortality from cardiovascular events, the extent of their adverse effects is not clear. This study explored the association between prescription of lipid lowering drugs and the risk of peripheral neuropathy. Design: A population based case-control study was carried out by linkage of several automated databases. Setting: Resident population of a northern Italian Province aged 40 years or more. Participants: Cases were patients discharged for peripheral neuropathy in 1998–1999. For each case up See end of article for authors’ affiliations to 20 controls were randomly selected among those eligible. Altogether 2040 case patients and 36 041 ....................... controls were included in the study. Exposure ascertainment: Prescription drug database was used to assess exposure to lipid lowering drugs Correspondence to: Professor G Corrao, at any time in the one year period preceding the index date. -
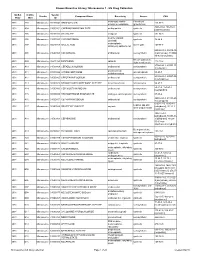
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM